Siegwald Léa, Touzet Hélène, Lemoine Yves, Hot David, Audebert Christophe, Caboche Ségolène
Gènes Diffusion, Douai, France.
CRIStAL (UMR CNRS 9189 University of Lille, Centre de Recherche en Informatique, Signal et Automatique de Lille) and Inria, Villeneuve d'Ascq, France.
PLoS One. 2017 Jan 4;12(1):e0169563. doi: 10.1371/journal.pone.0169563. eCollection 2017.
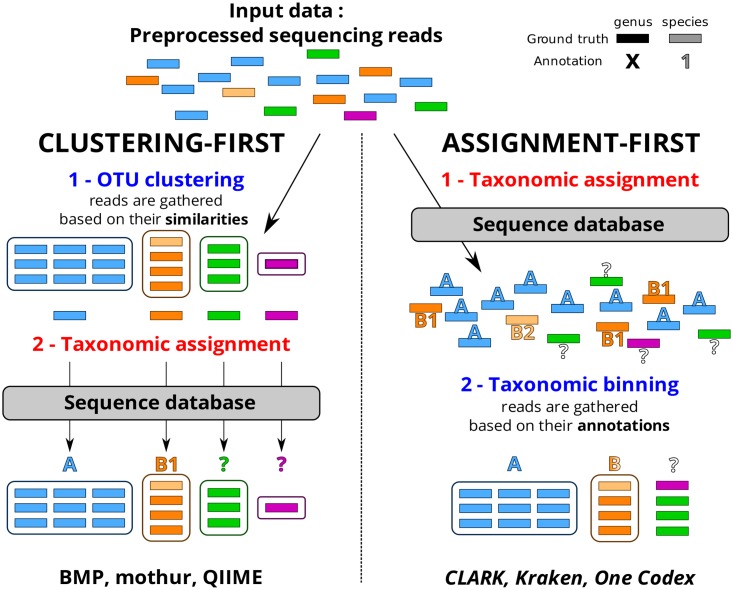
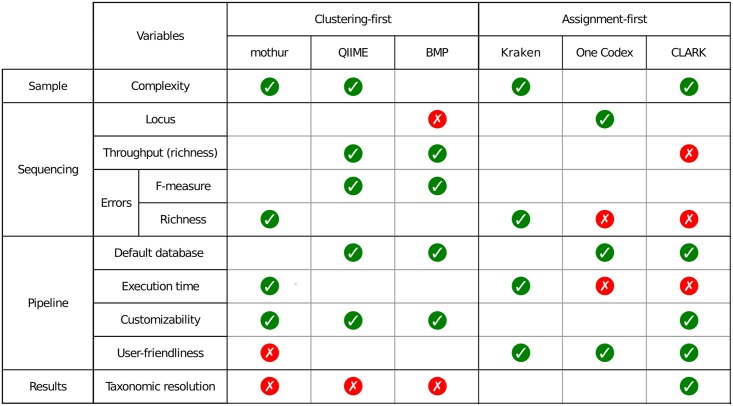
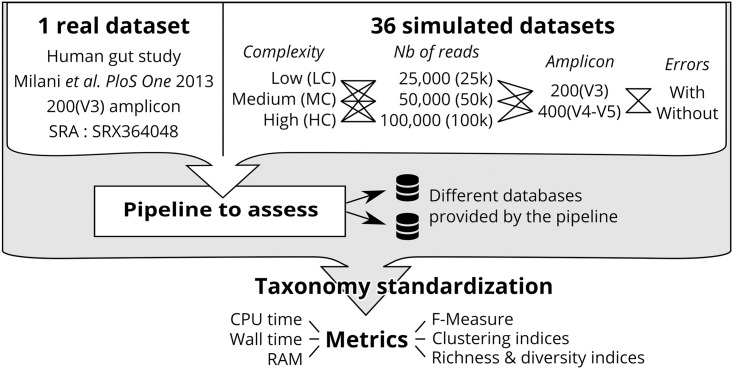
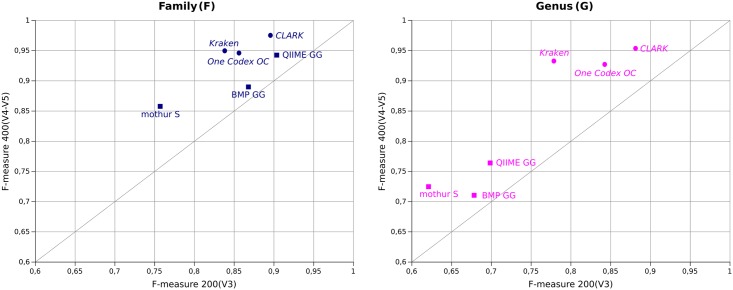
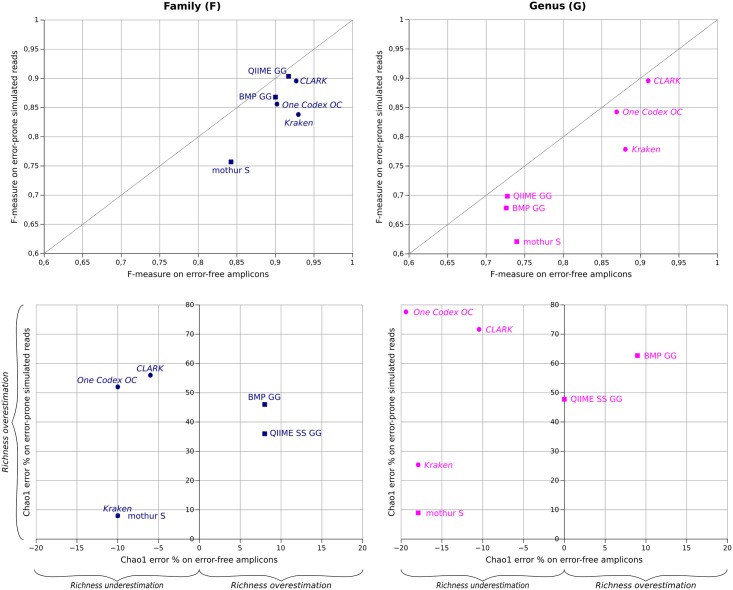
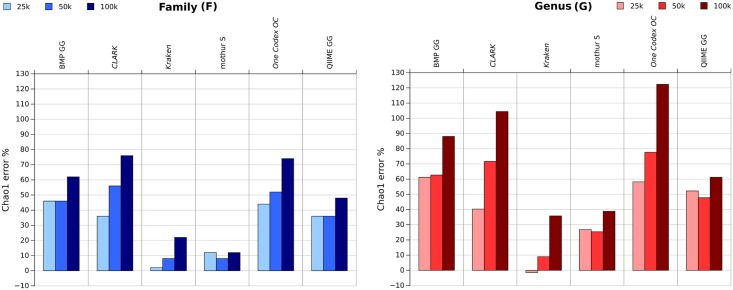
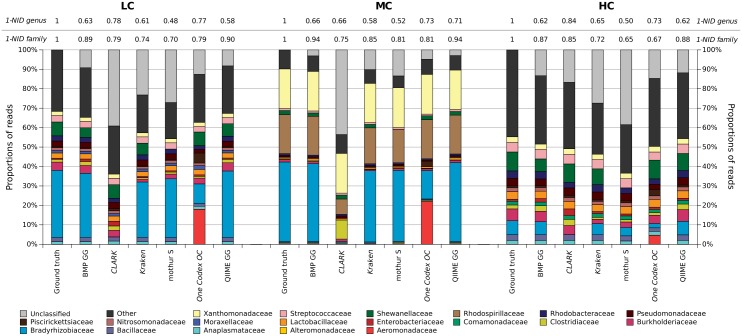
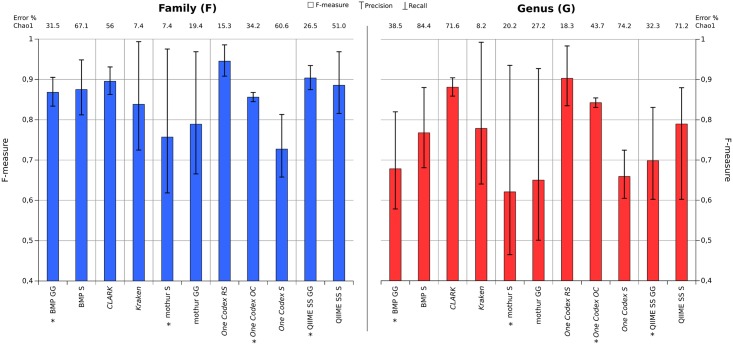
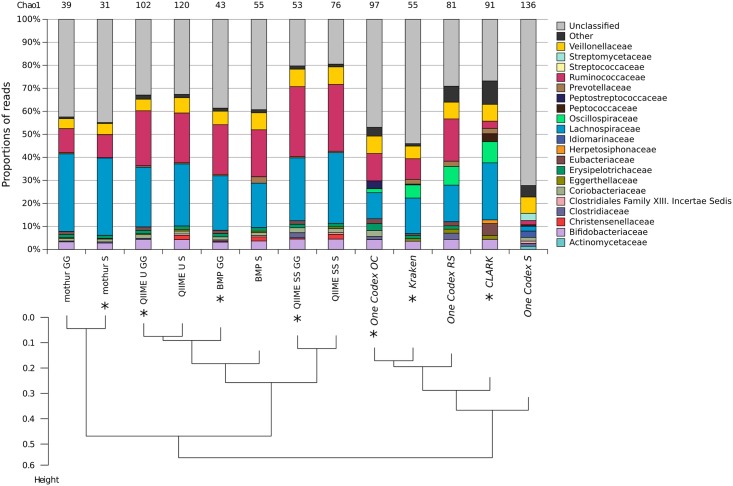
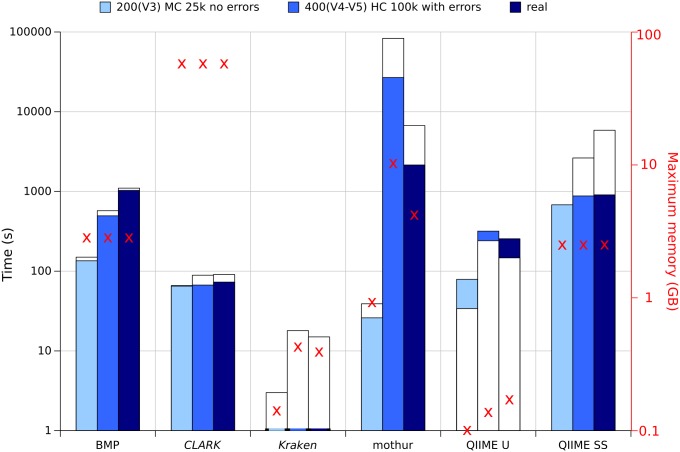
Targeted metagenomics, also known as metagenetics, is a high-throughput sequencing application focusing on a nucleotide target in a microbiome to describe its taxonomic content. A wide range of bioinformatics pipelines are available to analyze sequencing outputs, and the choice of an appropriate tool is crucial and not trivial. No standard evaluation method exists for estimating the accuracy of a pipeline for targeted metagenomics analyses. This article proposes an evaluation protocol containing real and simulated targeted metagenomics datasets, and adequate metrics allowing us to study the impact of different variables on the biological interpretation of results. This protocol was used to compare six different bioinformatics pipelines in the basic user context: Three common ones (mothur, QIIME and BMP) based on a clustering-first approach and three emerging ones (Kraken, CLARK and One Codex) using an assignment-first approach. This study surprisingly reveals that the effect of sequencing errors has a bigger impact on the results that choosing different amplified regions. Moreover, increasing sequencing throughput increases richness overestimation, even more so for microbiota of high complexity. Finally, the choice of the reference database has a bigger impact on richness estimation for clustering-first pipelines, and on correct taxa identification for assignment-first pipelines. Using emerging assignment-first pipelines is a valid approach for targeted metagenomics analyses, with a quality of results comparable to popular clustering-first pipelines, even with an error-prone sequencing technology like Ion Torrent. However, those pipelines are highly sensitive to the quality of databases and their annotations, which makes clustering-first pipelines still the only reliable approach for studying microbiomes that are not well described.
靶向宏基因组学,也被称为宏遗传学,是一种高通量测序应用,专注于微生物组中的核苷酸靶标以描述其分类内容。有多种生物信息学流程可用于分析测序输出结果,选择合适的工具至关重要且并非易事。目前不存在用于估计靶向宏基因组学分析流程准确性的标准评估方法。本文提出了一种评估方案,其中包含真实和模拟的靶向宏基因组学数据集,以及适当的指标,使我们能够研究不同变量对结果生物学解释的影响。该方案用于在基本用户环境中比较六种不同的生物信息学流程:三种基于先聚类方法的常用流程( mothur、QIIME 和 BMP)以及三种使用先分配方法的新兴流程(Kraken、CLARK 和 One Codex)。这项研究惊人地发现,测序错误的影响对结果的影响比对选择不同扩增区域的影响更大。此外,增加测序通量会增加丰度高估,对于高复杂性微生物群更是如此。最后,参考数据库的选择对基于先聚类流程的丰度估计影响更大,而对基于先分配流程的正确分类群鉴定影响更大。使用新兴的先分配流程是靶向宏基因组学分析的一种有效方法,其结果质量与流行的先聚类流程相当,即使使用像 Ion Torrent 这样容易出错的测序技术也是如此。然而,这些流程对数据库及其注释的质量高度敏感,这使得先聚类流程仍然是研究描述不充分的微生物组的唯一可靠方法。