El-Araby Moustafa E, Omar Abdelsattar M, Khayat Maan T, Assiri Hanan A, Al-Abd Ahmed M
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, King Abdulaziz University, Jeddah, Saudi Arabia.
Department of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Helwan University, Cairo, Egypt.
PLoS One. 2017 Jan 9;12(1):e0168938. doi: 10.1371/journal.pone.0168938. eCollection 2017.
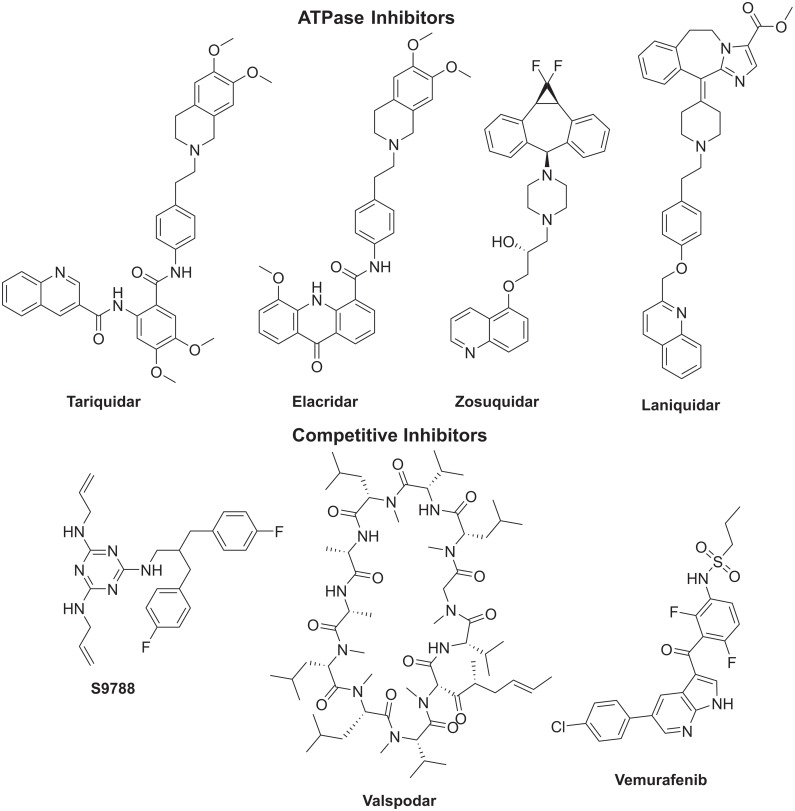
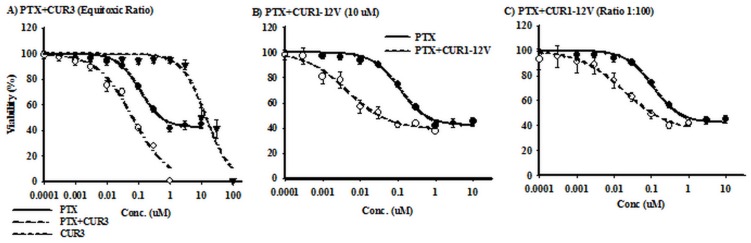
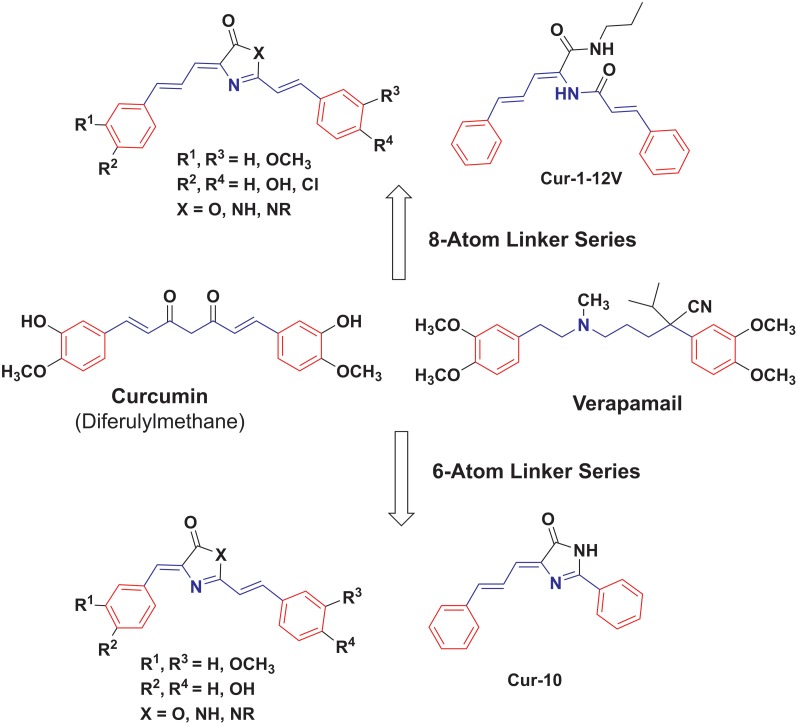
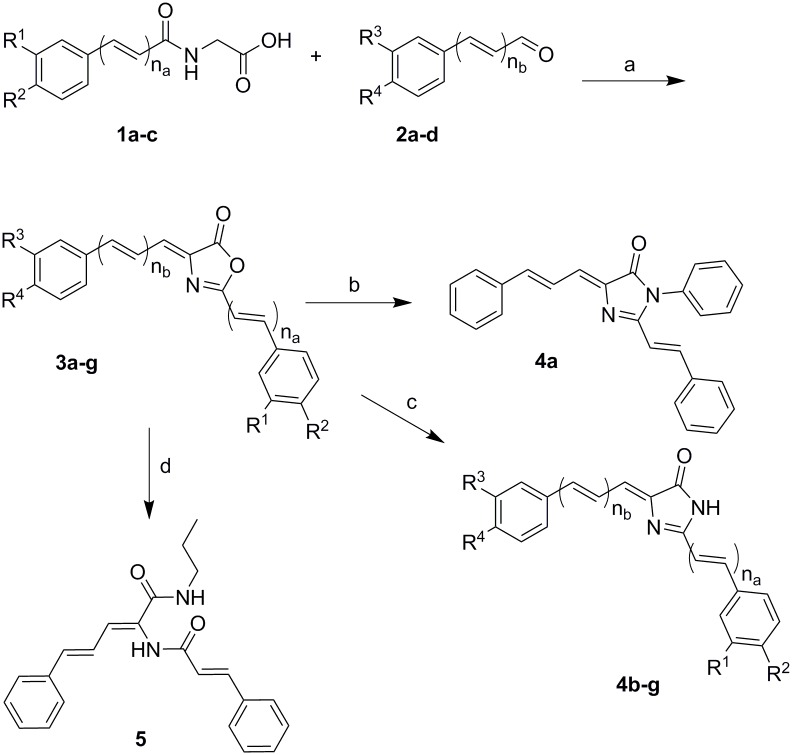
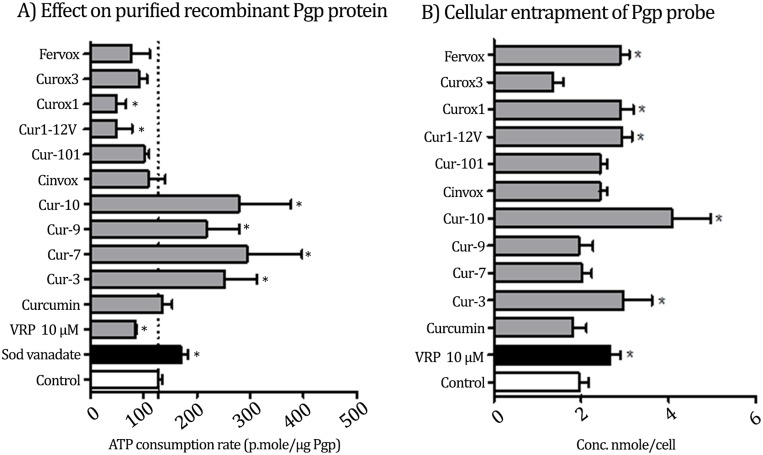
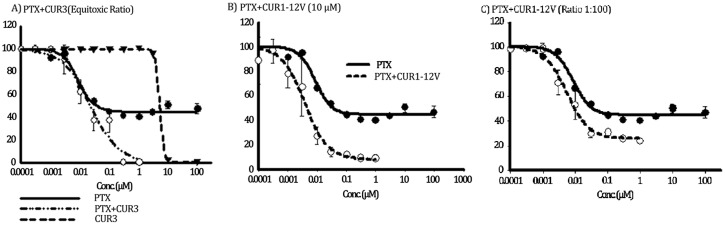
P-glycoprotein (Pgp) is a membrane bound efflux pump spread in a variety of tumor cells and considered as a main component of multidrug resistance (MDR) to chemotherapies. In this work, three groups of compounds (imidazolone, oxazolone and vinyl dipeptide derivatives) were synthesized aiming to develop a molecular framework that effectively suppresses MDR. When tested for their influence on Pgp activity, four compounds coded Cur1-01, Cur1-12V, Curox-1 and Curox-3 significantly decreased remaining ATP concentration indicating Pgp substrate site blocking. On the other hand, Cur-3 and Cur-10 significantly increased remaining ATP concentration, which is indicative of Pgp ATPase inhibition. The cytotoxicity of synthesized compounds was examined against Pgp expressing/highly resistant colorectal cancer cell lines (LS-174T). Compounds Cur-1 and Cur-3 showed considerable cytotoxicity with IC50 values of 7.6 and 8.9 μM, respectively. Equitoxic combination (at IC50 concentrations) of PTX and Cur-3 greatly diminished resistant cell clone from 45.7% to 2.5%, albeit with some drop in potency from IC50 of 7.9 nM to IC50 of 23.8 nM. On the other hand, combination of PTX and the non-cytotoxic Cur1-12V (10 μM) significantly decreased the IC50 of PTX to 3.8 nM as well as the resistant fraction to 16.2%. The combination test was confirmed using the same protocol but on another resistant CRC cell line (HCT-116) as we obtained similar results. Both Cur-3 and Cur1-12V (10 μM) significantly increased the cellular entrapment of Pgp probe (doxorubicin) elevating its intracellular concentration from 1.9 pmole/cell to 3.0 and 2.9 pmole/cell, respectively.
P-糖蛋白(Pgp)是一种膜结合外排泵,存在于多种肿瘤细胞中,被认为是化疗多药耐药性(MDR)的主要成分。在这项研究中,合成了三组化合物(咪唑啉酮、恶唑酮和乙烯基二肽衍生物),旨在开发一种能有效抑制多药耐药性的分子框架。在测试它们对Pgp活性的影响时,四种编码为Cur1-01、Cur1-12V、Curox-1和Curox-3的化合物显著降低了剩余ATP浓度,表明Pgp底物位点被阻断。另一方面,Cur-3和Cur-10显著提高了剩余ATP浓度,这表明Pgp ATP酶受到抑制。检测了合成化合物对表达Pgp/高耐药性结肠癌细胞系(LS-174T)的细胞毒性。化合物Cur-1和Cur-3表现出相当大的细胞毒性,IC50值分别为7.6和8.9μM。PTX和Cur-3的等效毒性组合(在IC50浓度下)使耐药细胞克隆从45.7%大幅减少至2.5%,尽管效力有所下降,IC50从7.9 nM降至23.8 nM。另一方面,PTX与无细胞毒性的Cur1-12V(10μM)组合显著降低了PTX的IC50至3.8 nM,同时耐药分数降至16.2%。使用相同方案在另一种耐药性结直肠癌细胞系(HCT-116)上进行组合测试,得到了类似结果。Cur-3和Cur1-12V(10μM)均显著增加了Pgp探针(阿霉素)的细胞内潴留,使其细胞内浓度分别从1.9皮摩尔/细胞提高到3.0和2.9皮摩尔/细胞。