Sethi Suresh A, Linden Daniel, Wenburg John, Lewis Cara, Lemons Patrick, Fuller Angela, Hare Matthew P
US Geological Survey, New York Cooperative Fish and Wildlife Research Unit , Cornell University , Ithaca, NY 14853 , USA.
New York Cooperative Fish and Wildlife Research Unit , Department of Natural Resources, Cornell University , Ithaca, NY , USA.
R Soc Open Sci. 2016 Dec 21;3(12):160457. doi: 10.1098/rsos.160457. eCollection 2016 Dec.
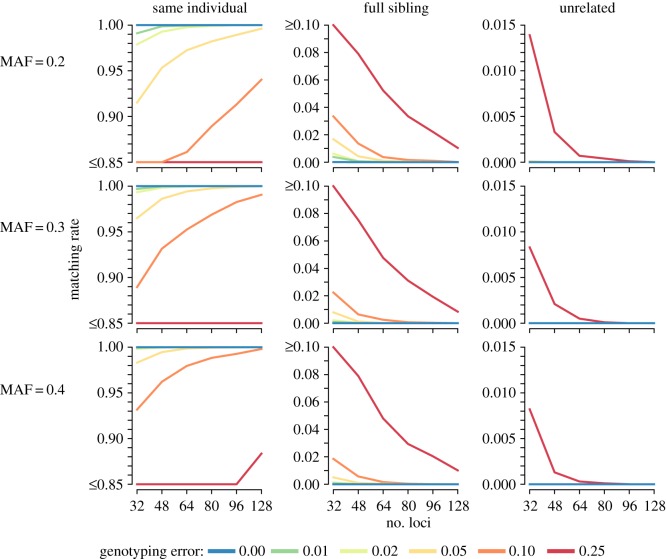
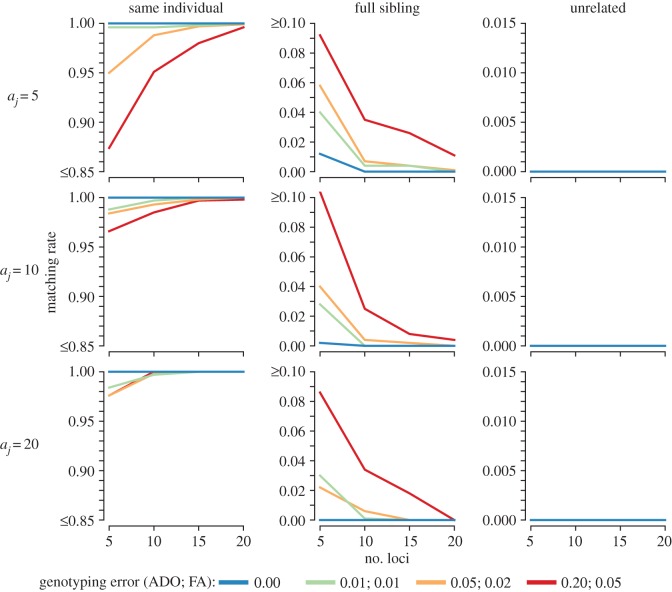
Error-tolerant likelihood-based match calling presents a promising technique to accurately identify recapture events in genetic mark-recapture studies by combining probabilities of latent genotypes and probabilities of observed genotypes, which may contain genotyping errors. Combined with clustering algorithms to group samples into sets of recaptures based upon pairwise match calls, these tools can be used to reconstruct accurate capture histories for mark-recapture modelling. Here, we assess the performance of a recently introduced error-tolerant likelihood-based match-calling model and sample clustering algorithm for genetic mark-recapture studies. We assessed both biallelic (i.e. single nucleotide polymorphisms; SNP) and multiallelic (i.e. microsatellite; MSAT) markers using a combination of simulation analyses and case study data on Pacific walrus () and fishers (). A novel two-stage clustering approach is demonstrated for genetic mark-recapture applications. First, repeat captures within a sampling occasion are identified. Subsequently, recaptures across sampling occasions are identified. The likelihood-based matching protocol performed well in simulation trials, demonstrating utility for use in a wide range of genetic mark-recapture studies. Moderately sized SNP (64+) and MSAT (10-15) panels produced accurate match calls for recaptures and accurate non-match calls for samples from closely related individuals in the face of low to moderate genotyping error. Furthermore, matching performance remained stable or increased as the number of genetic markers increased, genotyping error notwithstanding.
基于似然性的容错匹配调用提出了一种很有前景的技术,通过结合潜在基因型的概率和观察到的基因型的概率(其中可能包含基因分型错误),在遗传标记重捕研究中准确识别重捕事件。结合聚类算法,根据成对匹配调用将样本分组为重捕集,这些工具可用于为重捕建模重建准确的捕获历史。在这里,我们评估了一种最近引入的基于似然性的容错匹配调用模型和样本聚类算法在遗传标记重捕研究中的性能。我们使用模拟分析和关于太平洋海象()和渔貂()的案例研究数据的组合,评估了双等位基因(即单核苷酸多态性;SNP)和多等位基因(即微卫星;MSAT)标记。展示了一种用于遗传标记重捕应用的新颖的两阶段聚类方法。首先,识别采样期内的重复捕获。随后,识别跨采样期的重捕。基于似然性的匹配协议在模拟试验中表现良好,证明了其在广泛的遗传标记重捕研究中的实用性。面对低至中等的基因分型错误,中等大小的SNP(64个以上)和MSAT(10 - 15个)面板为重捕产生了准确的匹配调用,为来自密切相关个体的样本产生了准确的非匹配调用。此外,尽管存在基因分型错误,但随着遗传标记数量的增加,匹配性能保持稳定或提高。