Hu Yanmei, Musharrafieh Rami, Ma Chunlong, Zhang Jiantao, Smee Donald F, DeGrado William F, Wang Jun
Department of Pharmacology and Toxicology, College of Pharmacy, The University of Arizona, Tucson, Arizona 85721, United States.
Department of Chemistry and Biochemistry, The University of Arizona, Tucson, Arizona 85721, United States.
Antiviral Res. 2017 Apr;140:45-54. doi: 10.1016/j.antiviral.2017.01.006. Epub 2017 Jan 10.
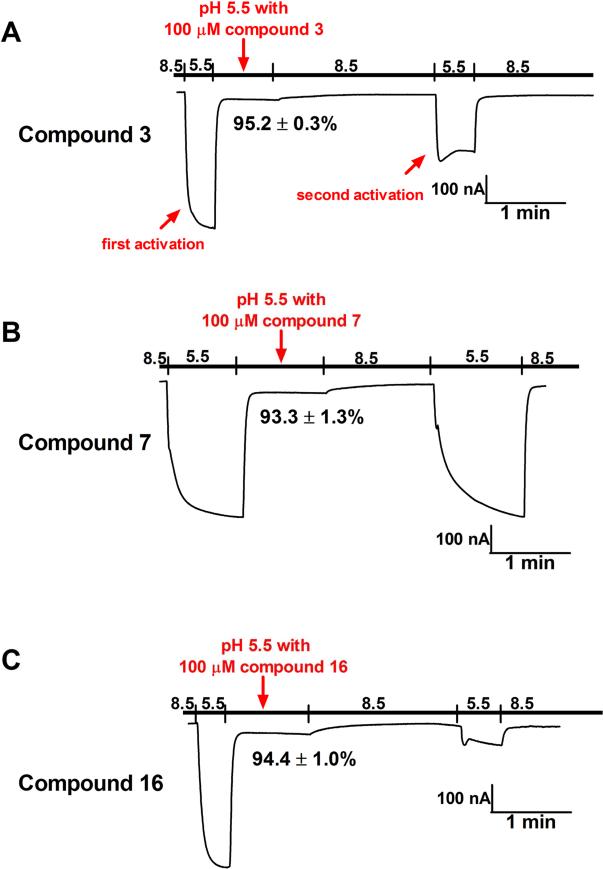
Adamantanes such as amantadine (1) and rimantadine (2) are FDA-approved anti-influenza drugs that act by inhibiting the wild-type M2 proton channel from influenza A viruses, thereby inhibiting the uncoating of the virus. Although adamantanes have been successfully used for more than four decades, their efficacy was curtailed by emerging drug resistance. Among the limited number of M2 mutants that confer amantadine resistance, the M2-V27A mutant was found to be the predominant mutant under drug selection pressure, thereby representing a high profile antiviral drug target. Guided by molecular dynamics simulations, we previously designed first-in-class M2-V27A inhibitors. One of the potent lead compounds, spiroadamantane amine (3), inhibits both the M2-WT and M2-V27A mutant with IC values of 18.7 and 0.3 μM, respectively, in in vitro electrophysiological assays. Encouraged by these findings, in this study we further examine the in vitro and in vivo antiviral activity of compound 3 in inhibiting both amantadine-sensitive and -resistant influenza A viruses. Compound 3 not only had single to sub-micromolar EC values against M2-WT- and M2-V27A-containing influenza A viruses in antiviral assays, but also rescued mice from lethal viral infection by either M2-WT- or M2-V27A-containing influenza A viruses. In addition, we report the design of two analogs of compound 3, and one was found to have improved in vitro antiviral activity over compound 3. Collectively, this study represents the first report demonstrating the in vivo antiviral efficacy of inhibitors targeting M2 mutants. The results suggest that inhibitors targeting drug-resistant M2 mutants are promising antiviral drug candidates worthy of further development.
金刚烷类药物如金刚烷胺(1)和金刚乙胺(2)是美国食品药品监督管理局(FDA)批准的抗流感药物,其作用机制是抑制甲型流感病毒的野生型M2质子通道,从而抑制病毒脱壳。尽管金刚烷类药物已成功使用了四十多年,但新出现的耐药性使其疗效受到影响。在赋予金刚烷胺耐药性的有限数量的M2突变体中,M2-V27A突变体在药物选择压力下被发现是主要突变体,因此成为备受关注的抗病毒药物靶点。在分子动力学模拟的指导下,我们之前设计了一流的M2-V27A抑制剂。其中一种有效的先导化合物,螺金刚烷胺(3),在体外电生理试验中分别以18.7和0.3μM的IC值抑制M2-WT和M2-V27A突变体。受这些发现的鼓舞,在本研究中我们进一步研究了化合物3在体外和体内对金刚烷胺敏感和耐药甲型流感病毒的抗病毒活性。化合物3不仅在抗病毒试验中对含M2-WT和M2-V27A的甲型流感病毒具有单微摩尔至亚微摩尔的EC值,而且还能使感染含M2-WT或M2-V27A的甲型流感病毒的小鼠免于致命的病毒感染。此外,我们报告了化合物3的两种类似物的设计,其中一种在体外抗病毒活性方面比化合物3有所提高。总的来说,本研究是首次证明靶向M2突变体的抑制剂具有体内抗病毒疗效的报告。结果表明,靶向耐药M2突变体的抑制剂是有前景的抗病毒药物候选物,值得进一步开发。