Maringer Kevin, Yousuf Amjad, Heesom Kate J, Fan Jun, Lee David, Fernandez-Sesma Ana, Bessant Conrad, Matthews David A, Davidson Andrew D
School of Cellular and Molecular Medicine, University of Bristol, Bristol, BS8 1TD, UK.
Department of Microbiology, Icahn School of Medicine at Mount Sinai, New York, 10029, NY, USA.
BMC Genomics. 2017 Jan 19;18(1):101. doi: 10.1186/s12864-016-3432-5.
Aedes aegypti is a vector for the (re-)emerging human pathogens dengue, chikungunya, yellow fever and Zika viruses. Almost half of the Ae. aegypti genome is comprised of transposable elements (TEs). Transposons have been linked to diverse cellular processes, including the establishment of viral persistence in insects, an essential step in the transmission of vector-borne viruses. However, up until now it has not been possible to study the overall proteome derived from an organism's mobile genetic elements, partly due to the highly divergent nature of TEs. Furthermore, as for many non-model organisms, incomplete genome annotation has hampered proteomic studies on Ae. aegypti.
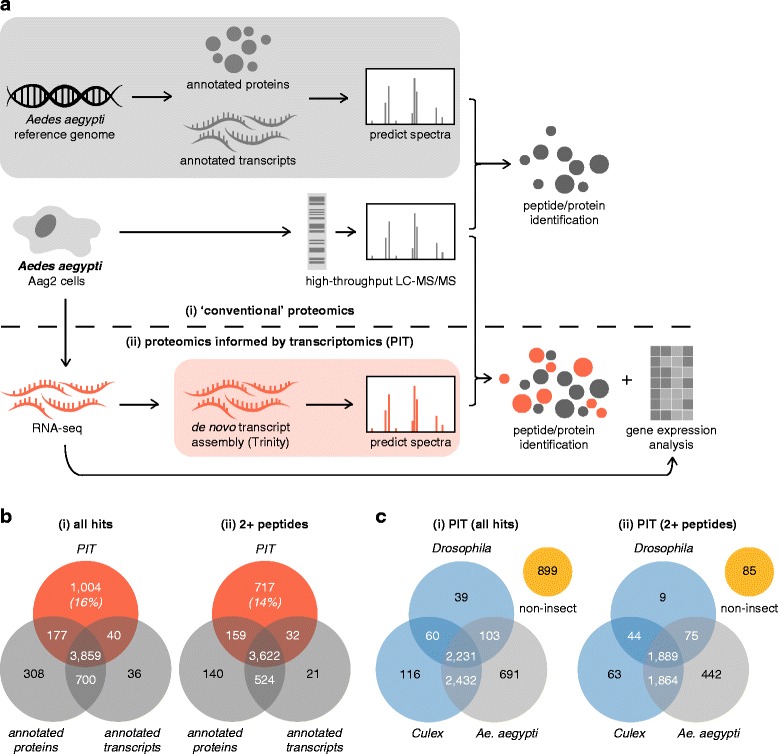
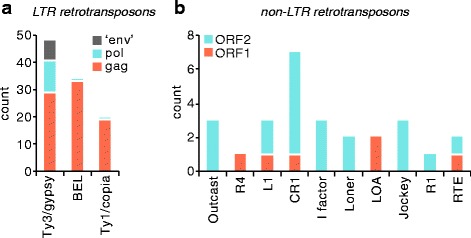
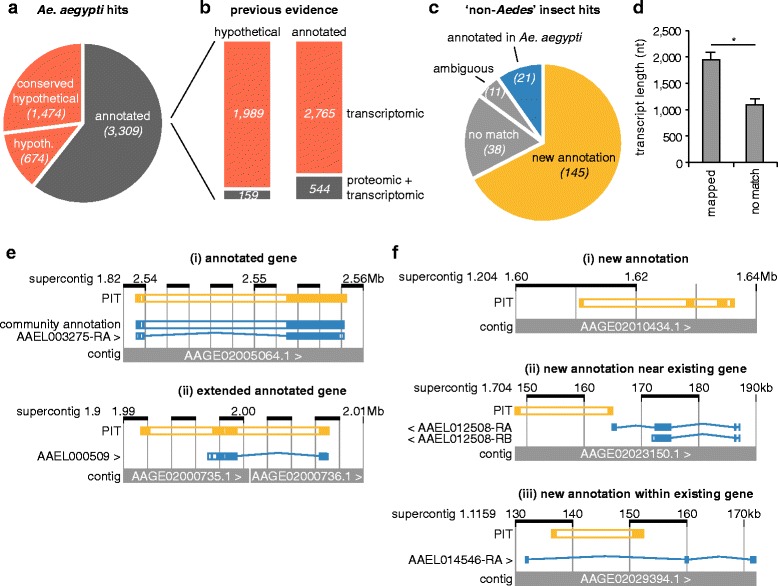
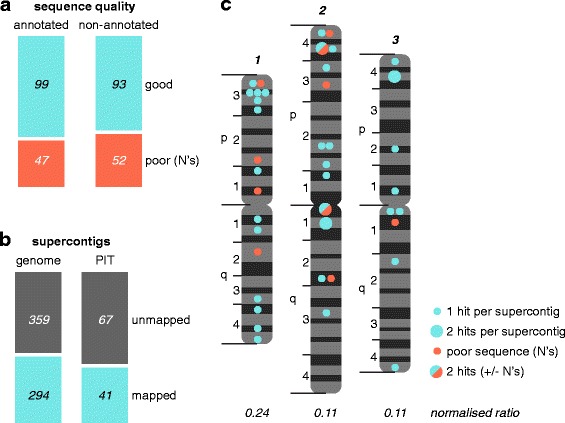
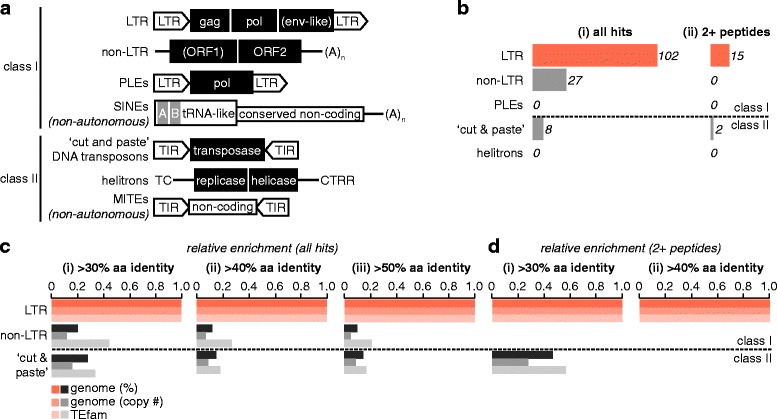
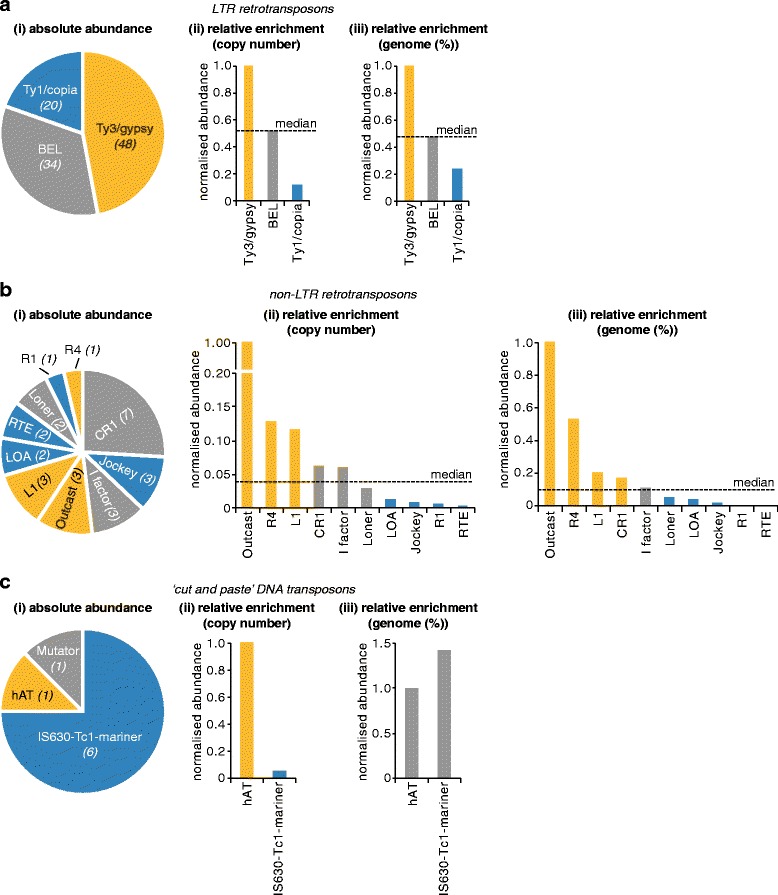
We analysed the Ae. aegypti proteome using our new proteomics informed by transcriptomics (PIT) technique, which bypasses the need for genome annotation by identifying proteins through matched transcriptomic (rather than genomic) data. Our data vastly increase the number of experimentally confirmed Ae. aegypti proteins. The PIT analysis also identified hotspots of incomplete genome annotation, and showed that poor sequence and assembly quality do not explain all annotation gaps. Finally, in a proof-of-principle study, we developed criteria for the characterisation of proteomically active TEs. Protein expression did not correlate with a TE's genomic abundance at different levels of classification. Most notably, long terminal repeat (LTR) retrotransposons were markedly enriched compared to other elements. PIT was superior to 'conventional' proteomic approaches in both our transposon and genome annotation analyses.
We present the first proteomic characterisation of an organism's repertoire of mobile genetic elements, which will open new avenues of research into the function of transposon proteins in health and disease. Furthermore, our study provides a proof-of-concept that PIT can be used to evaluate a genome's annotation to guide annotation efforts which has the potential to improve the efficiency of annotation projects in non-model organisms. PIT therefore represents a valuable new tool to study the biology of the important vector species Ae. aegypti, including its role in transmitting emerging viruses of global public health concern.
埃及伊蚊是登革热、基孔肯雅热、黄热病和寨卡病毒等(重新)出现的人类病原体的传播媒介。埃及伊蚊基因组几乎一半由转座元件(TEs)组成。转座子与多种细胞过程相关,包括病毒在昆虫体内的持续存在,这是虫媒病毒传播的关键步骤。然而,到目前为止,由于转座元件的高度多样性,还无法研究源自生物体移动遗传元件的整体蛋白质组。此外,对于许多非模式生物来说,基因组注释不完整阻碍了对埃及伊蚊的蛋白质组学研究。
我们使用新的转录组学指导蛋白质组学(PIT)技术分析了埃及伊蚊蛋白质组,该技术通过匹配的转录组(而非基因组)数据识别蛋白质,从而无需基因组注释。我们的数据极大地增加了经实验确认的埃及伊蚊蛋白质的数量。PIT分析还确定了基因组注释不完整的热点区域,并表明序列和组装质量差并不能解释所有注释缺口。最后,在一项原理验证研究中,我们制定了蛋白质组学活跃转座元件的表征标准。在不同分类水平上,蛋白质表达与转座元件的基因组丰度不相关。最显著的是,与其他元件相比,长末端重复(LTR)逆转座子明显富集。在转座子和基因组注释分析中,PIT均优于“传统”蛋白质组学方法。
我们首次对生物体的移动遗传元件库进行了蛋白质组学表征,这将为研究转座子蛋白在健康和疾病中的功能开辟新的研究途径。此外,我们的研究提供了一个概念验证,即PIT可用于评估基因组注释,以指导注释工作,这有可能提高非模式生物注释项目的效率。因此,PIT是研究重要病媒物种埃及伊蚊生物学的一种有价值的新工具,包括其在传播全球公共卫生关注的新兴病毒中的作用。