Si Dong, He Jing
Division of Computing and Software Systems, University of Washington Bothell, Bothell, WA 98011, USA.
Department of Computer Science, Old Dominion University, Norfolk, VA 23529, USA.
Biomed Res Int. 2017;2017:1793213. doi: 10.1155/2017/1793213. Epub 2017 Jan 10.
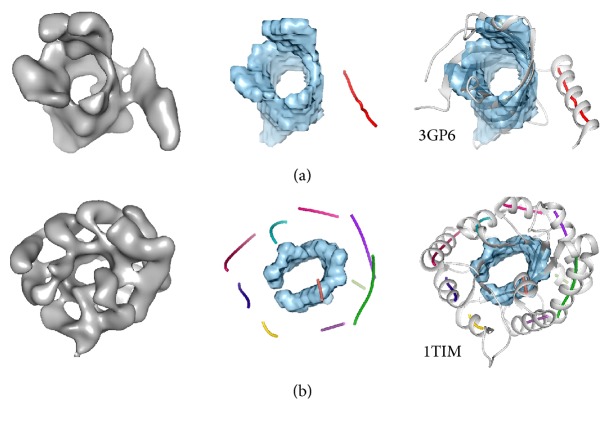
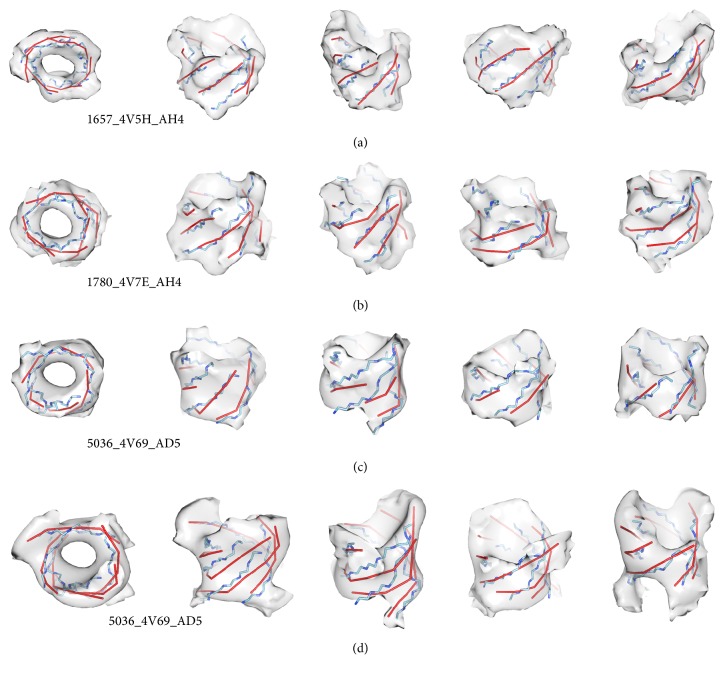
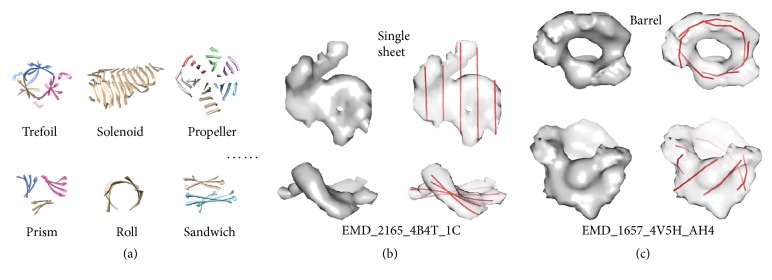
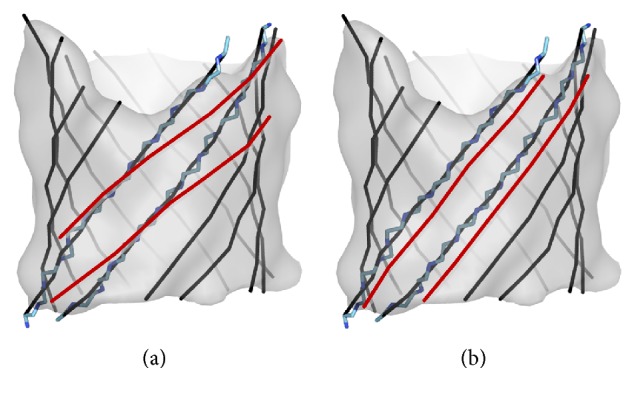
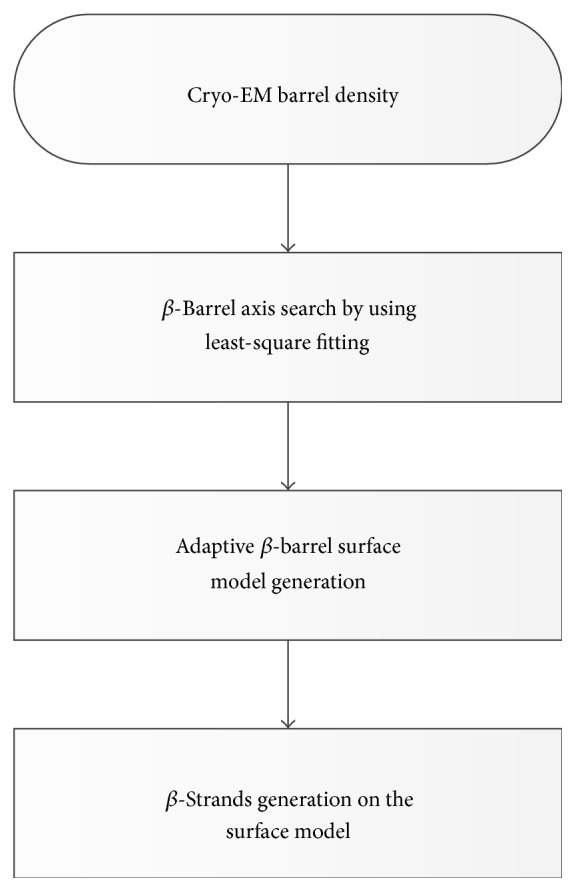
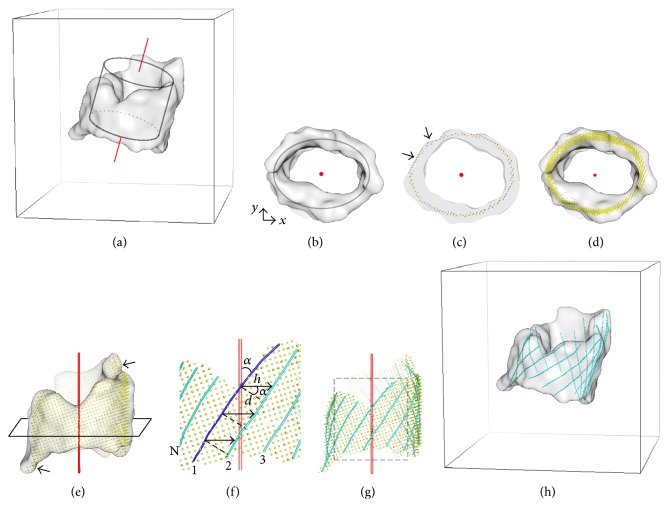
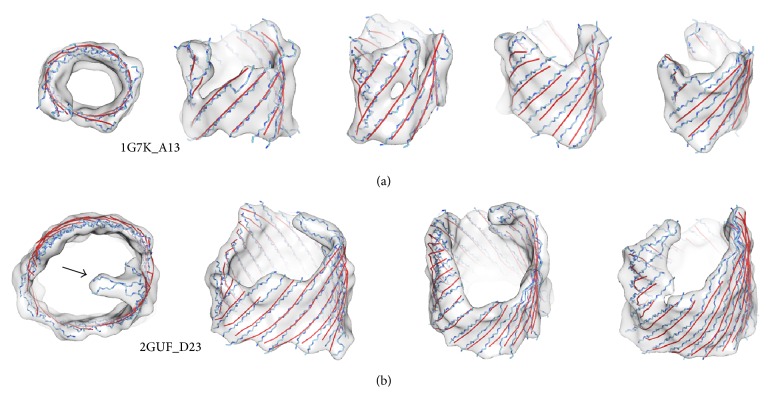
Cryo-electron microscopy (cryo-EM) has produced density maps of various resolutions. Although -helices can be detected from density maps at 5-8 Å resolutions, -strands are challenging to detect at such density maps due to close-spacing of -strands. The variety of shapes of -sheets adds the complexity of -strands detection from density maps. We propose a new approach to model traces of -strands for -barrel density regions that are extracted from cryo-EM density maps. In the test containing eight -barrels extracted from experimental cryo-EM density maps at 5.5 Å-8.25 Å resolution, detected about 74.26% of the amino acids in the -strands with an overall 2.05 Å 2-way distance between the detected -traces and the observed ones, if the best of the fifteen detection cases is considered.
冷冻电子显微镜(cryo-EM)已生成了各种分辨率的密度图。尽管在5-8埃分辨率的密度图中可以检测到α螺旋,但由于β链间距紧密,在这样的密度图中检测β链具有挑战性。β折叠的多种形状增加了从密度图中检测β链的复杂性。我们提出了一种新方法,用于对从冷冻电子显微镜密度图中提取的β桶密度区域的β链轨迹进行建模。在包含从5.5埃至8.25埃分辨率的实验冷冻电子显微镜密度图中提取的8个β桶的测试中,如果考虑15个检测案例中的最佳结果,检测到的β链中约74.26%的氨基酸,检测到的β轨迹与观察到的β轨迹之间的总体双向距离为2.05埃。