Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany.
Department of Earth Sciences, University College London, London WC1E 6BT, UK.
Nat Commun. 2017 Feb 7;8:14052. doi: 10.1038/ncomms14052.
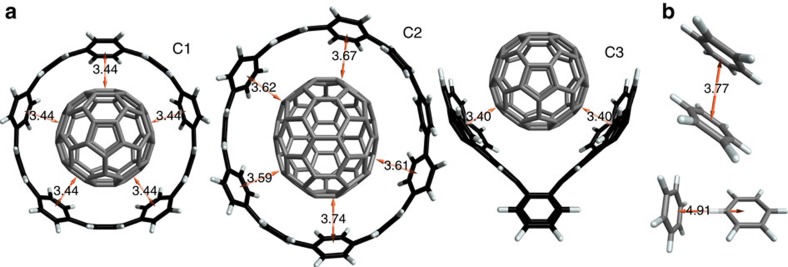
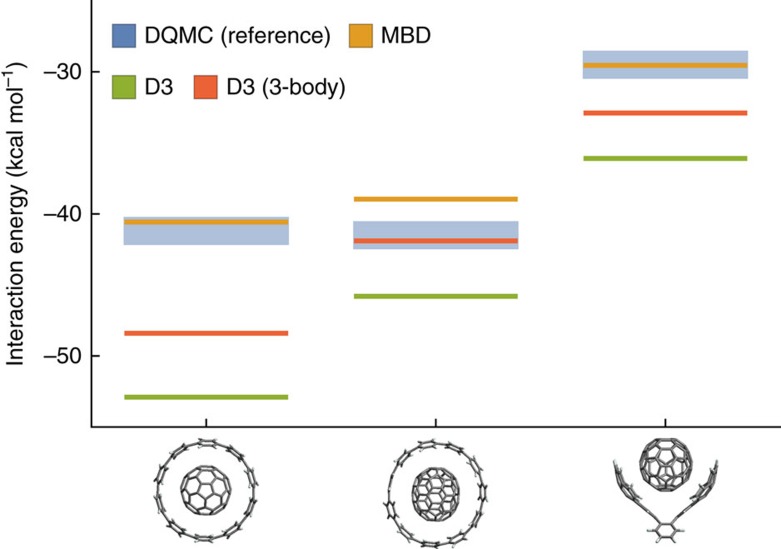
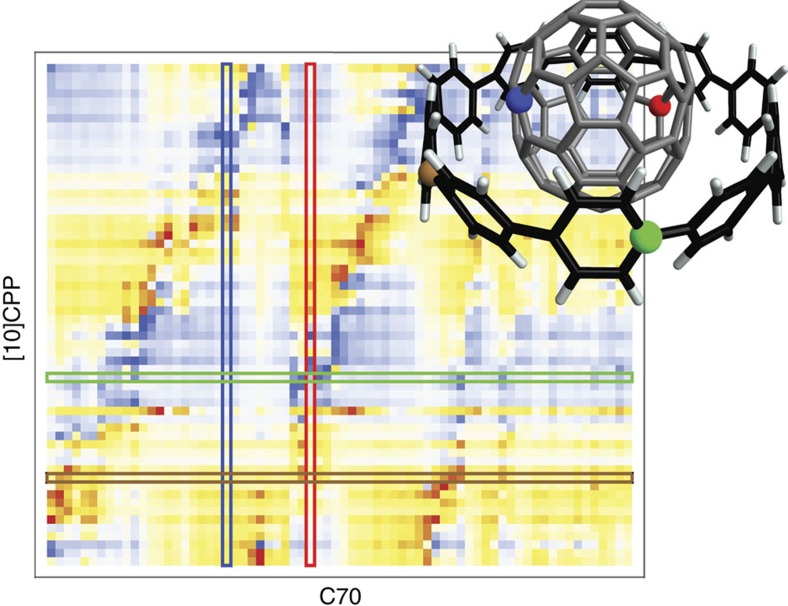
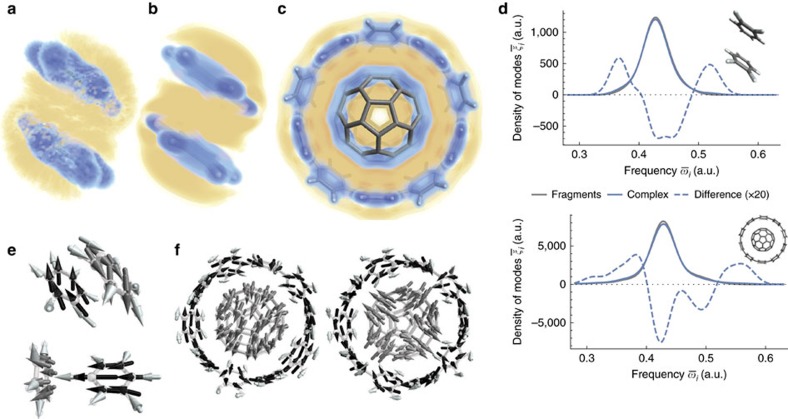
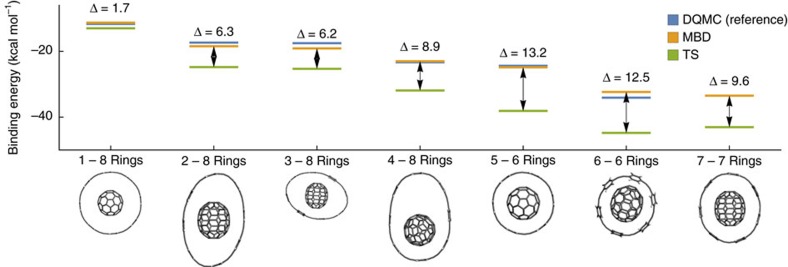
Non-covalent π-π interactions are central to chemical and biological processes, yet the full understanding of their origin that would unite the simplicity of empirical approaches with the accuracy of quantum calculations is still missing. Here we employ a quantum-mechanical Hamiltonian model for van der Waals interactions, to demonstrate that intermolecular electron correlation in large supramolecular complexes at equilibrium distances is appropriately described by collective charge fluctuations. We visualize these fluctuations and provide connections both to orbital-based approaches to electron correlation, as well as to the simple London pairwise picture. The reported binding energies of ten supramolecular complexes obtained from the quantum-mechanical fluctuation model joined with density functional calculations are within 5% of the reference energies calculated with the diffusion quantum Monte-Carlo method. Our analysis suggests that π-π stacking in supramolecular complexes can be characterized by strong contributions to the binding energy from delocalized, collective charge fluctuations-in contrast to complexes with other types of bonding.
非共价的 π-π 相互作用是化学和生物过程的核心,但要将经验方法的简单性与量子计算的准确性结合起来,全面理解其起源仍然缺失。在这里,我们采用了范德华相互作用的量子力学哈密顿模型,证明了在平衡距离处的大超分子复合物中的分子间电子相关可以通过集体电荷涨落得到适当描述。我们对这些涨落进行了可视化,并提供了与基于轨道的电子相关方法以及简单的伦敦对图像之间的联系。从量子力学涨落模型与密度泛函计算相结合得到的十个超分子复合物的结合能与用扩散量子蒙特卡罗方法计算的参考能相差在 5%以内。我们的分析表明,在超分子复合物中,π-π 堆积可以通过离域的、集体电荷涨落对结合能的强烈贡献来表征,这与具有其他类型键的复合物形成对比。