Wellcome Trust Centre for Human Genetics, OX3 7BN Oxford, UK.
Wellcome Trust Sanger Institute, Wellcome Genome Campus, CB10 1SA Hinxton, Cambridge, UK.
Sci Data. 2017 Feb 14;4:170011. doi: 10.1038/sdata.2017.11.
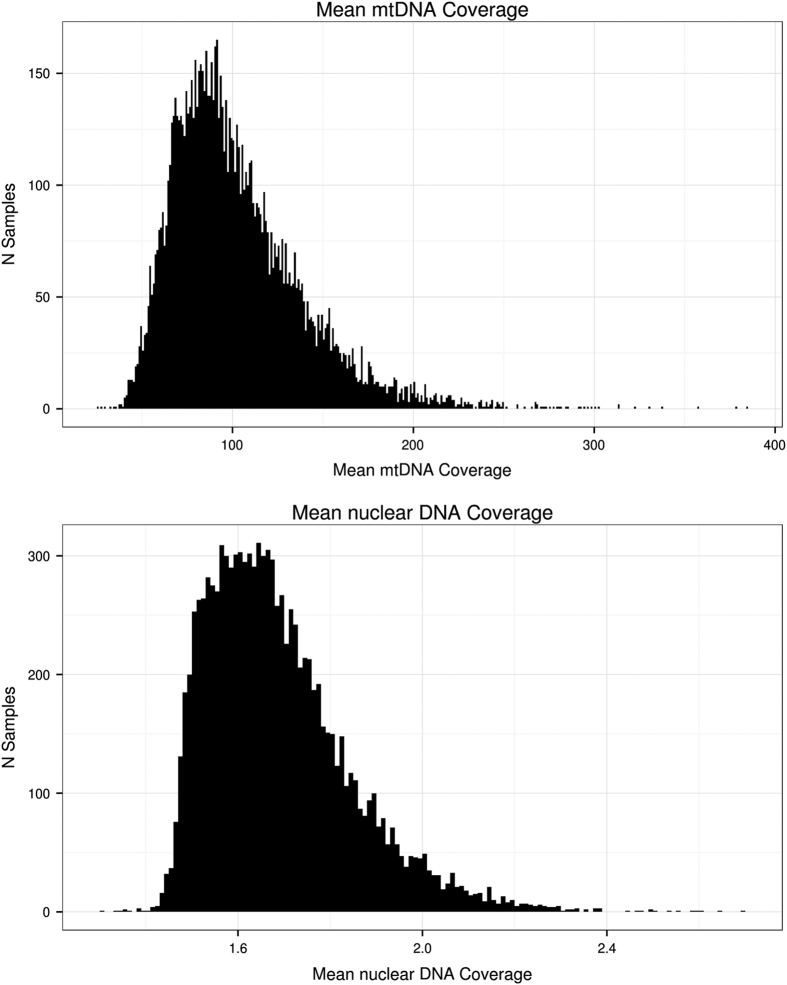
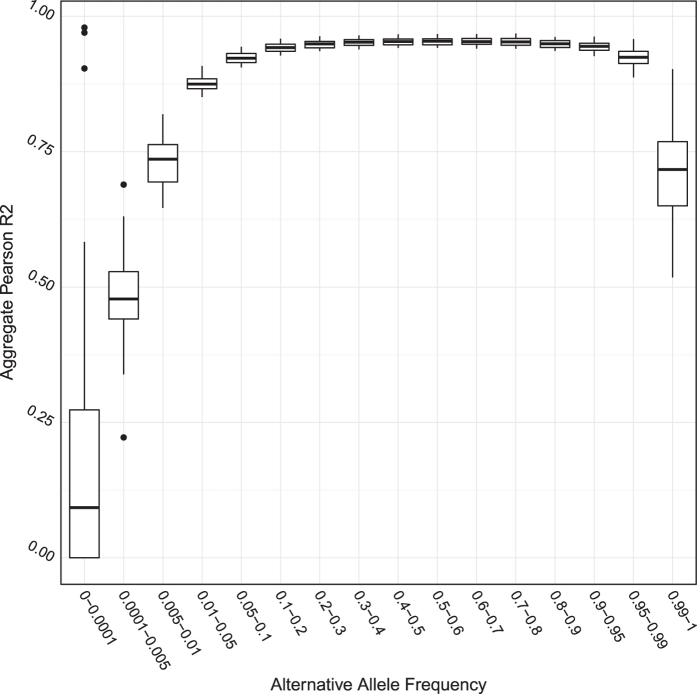
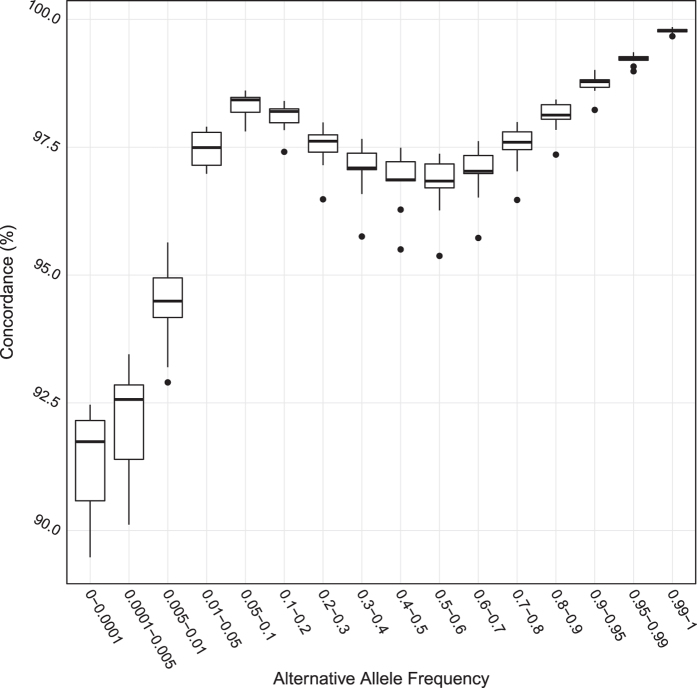
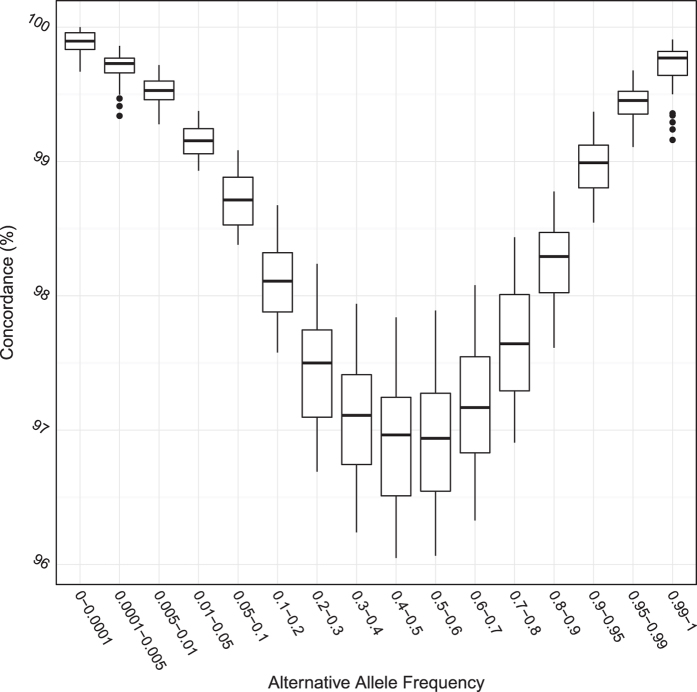
The China, Oxford and Virginia Commonwealth University Experimental Research on Genetic Epidemiology (CONVERGE) project on Major Depressive Disorder (MDD) sequenced 11,670 female Han Chinese at low-coverage (1.7X), providing the first large-scale whole genome sequencing resource representative of the largest ethnic group in the world. Samples are collected from 58 hospitals from 23 provinces around China. We are able to call 22 million high quality single nucleotide polymorphisms (SNP) from the nuclear genome, representing the largest SNP call set from an East Asian population to date. We use these variants for imputation of genotypes across all samples, and this has allowed us to perform a successful genome wide association study (GWAS) on MDD. The utility of these data can be extended to studies of genetic ancestry in the Han Chinese and evolutionary genetics when integrated with data from other populations. Molecular phenotypes, such as copy number variations and structural variations can be detected, quantified and analysed in similar ways.
中国、牛津大学和弗吉尼亚联邦大学的遗传流行病学(CONVERGE)项目对重度抑郁症(MDD)进行了研究,对 11670 名汉族女性进行了低覆盖率(1.7X)的测序,提供了世界上最大族群的首个大规模全基因组测序资源。样本来自中国 23 个省的 58 家医院。我们能够从核基因组中调用 2200 万个高质量的单核苷酸多态性(SNP),这是迄今为止来自东亚人群的最大 SNP 调用集。我们使用这些变体对所有样本进行基因型推断,这使我们能够成功地对 MDD 进行全基因组关联研究(GWAS)。当与其他人群的数据相结合时,这些数据的用途可以扩展到汉族遗传祖先和进化遗传学的研究中。可以以类似的方式检测、量化和分析分子表型,如拷贝数变异和结构变异。