Department of Epigenetics, Medical Research Institute, Tokyo Medical and Dental University (TMDU), Bunkyo-ku, Tokyo 113-8510, Japan.
Institute for Protein Research, Osaka University, Suita, Osaka 565-0871, Japan.
Nucleic Acids Res. 2017 Feb 28;45(4):e24. doi: 10.1093/nar/gkw994.
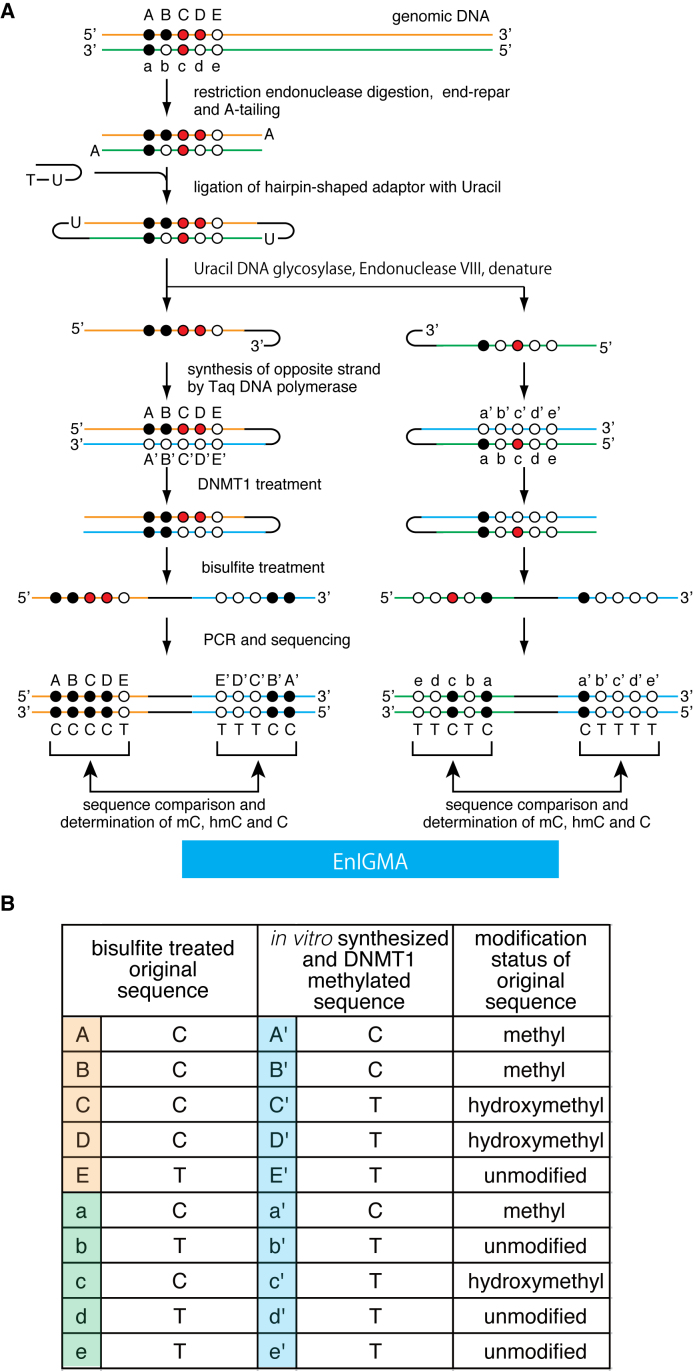
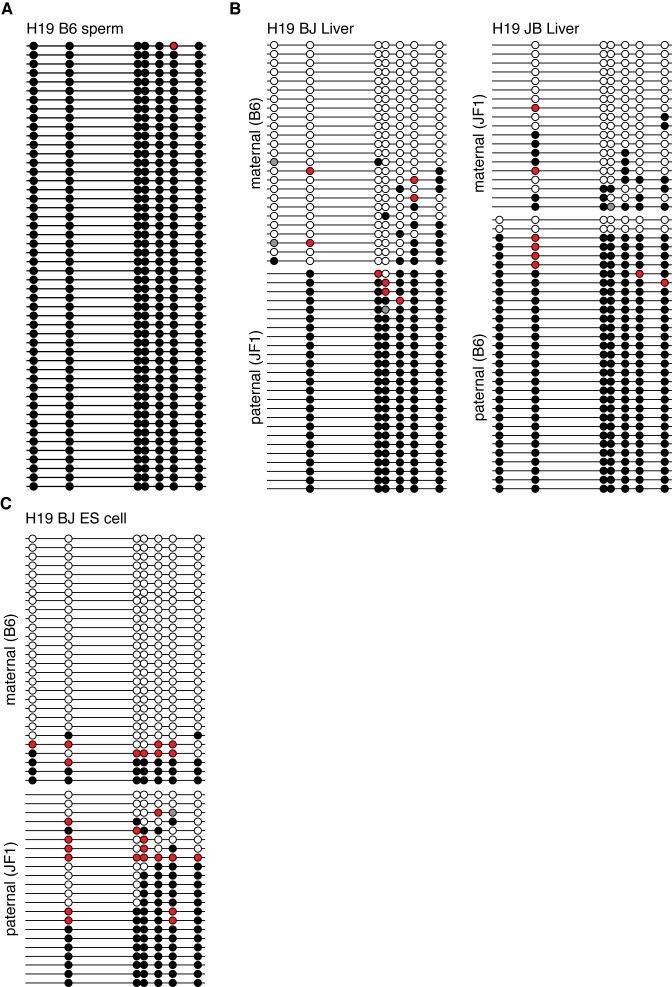
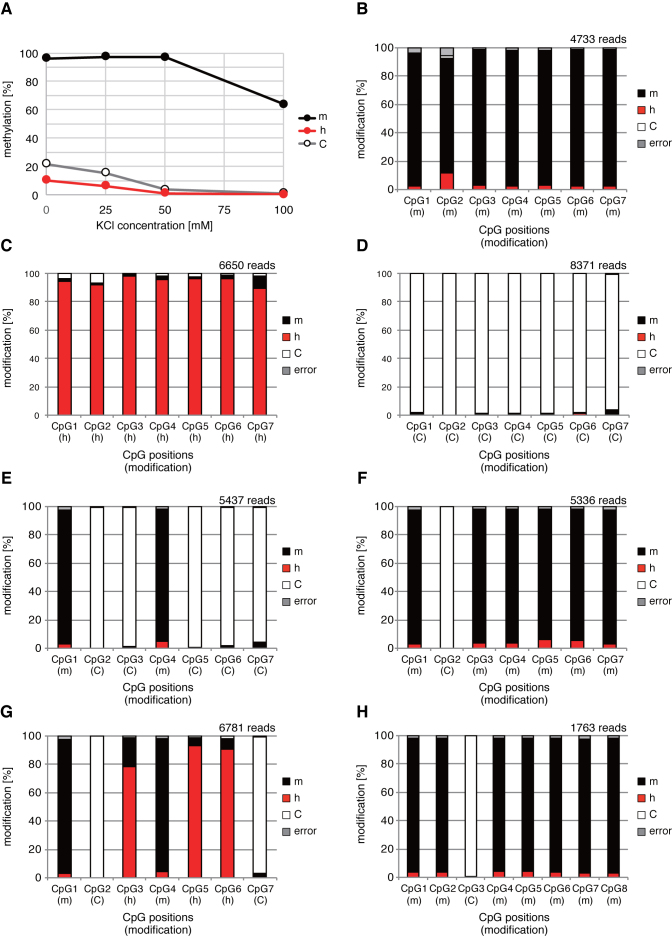
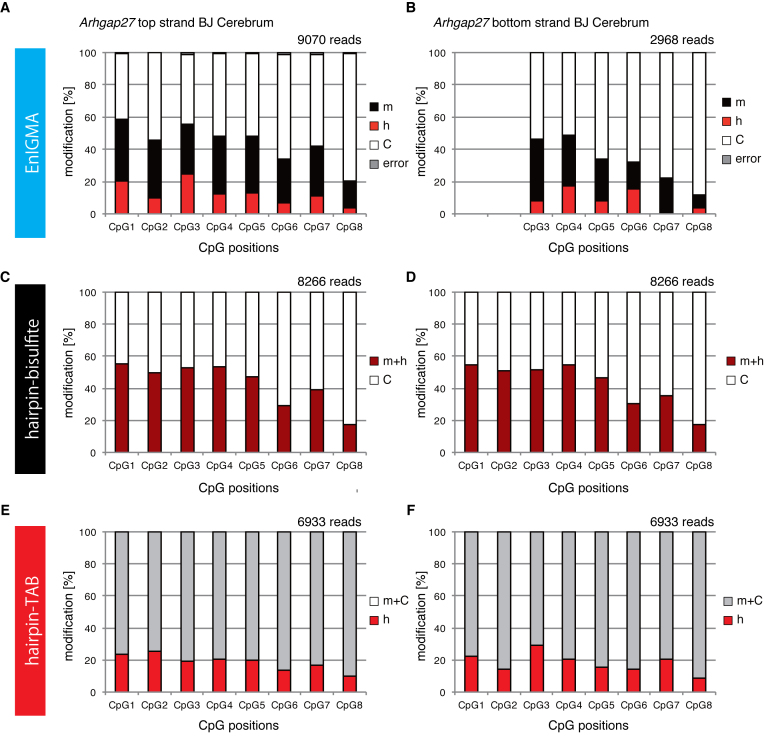
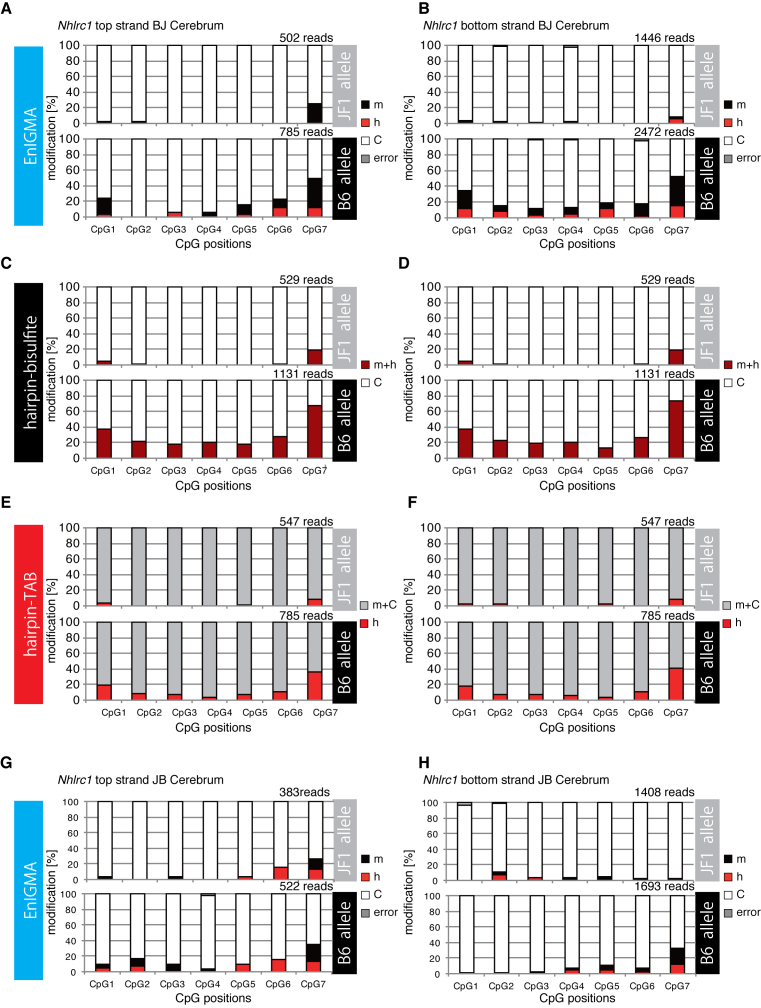
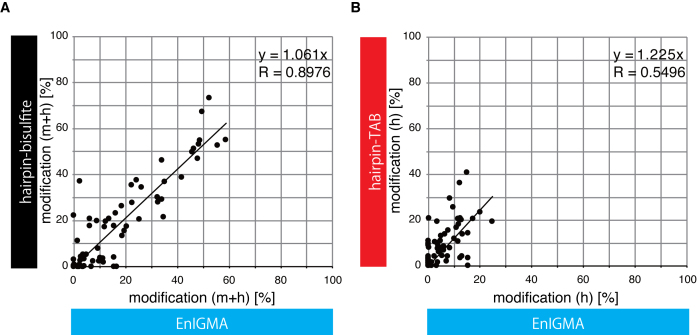
Since the discovery of oxidative demethylation of methylcytosine (mC) by Tet enzymes, an analytical method has been urgently needed that would enable the identification of mC and hydroxymethylcytosine (hmC) at the single base resolution level, because their roles in gene regulation are quite different from each other. However, the bisulfite sequencing method, the gold standard for DNA methylation analysis at present, does not distinguish them. Recently reported alternative methods, such as oxBS-seq and TAB-seq, are not even capable of determining mC and hmC simultaneously. Here, we report a novel method for the direct identification of mC, hmC and unmodified cytosine (C) at a single base resolution. We named this method the Enzyme-assisted Identification of Genome Modification Assay (EnIGMA), and it was demonstrated to indeed have a highly efficient and reliable analytic capacity for distinguishing them. We also successfully applied this novel method to the analysis of the maintenance of the DNA methylation status of imprinted H19-DMR. Importantly, hydroxymethylation plays an ambivalent role in the maintenance of the genome imprinting memory in parental genomes essential for normal development, shedding new light on the epigenetic regulation in ES cells.
自从 Tet 酶发现了甲基胞嘧啶(mC)的氧化去甲基化作用以来,人们迫切需要一种分析方法,能够在单碱基分辨率水平上识别 mC 和 5-羟甲基胞嘧啶(hmC),因为它们在基因调控中的作用彼此截然不同。然而,目前 DNA 甲基化分析的金标准——亚硫酸氢盐测序方法并不能区分它们。最近报道的替代方法,如 oxBS-seq 和 TAB-seq,甚至不能同时测定 mC 和 hmC。在这里,我们报告了一种用于直接识别 mC、hmC 和未修饰胞嘧啶(C)的新方法,我们将其命名为酶辅助基因组修饰分析(EnIGMA),并证明它确实具有高效、可靠的分析能力来区分它们。我们还成功地将这种新方法应用于印迹 H19-DMR 的 DNA 甲基化状态维持分析。重要的是,羟甲基化在维持正常发育所必需的亲本基因组中的基因组印记记忆方面起着双重作用,为 ES 细胞中的表观遗传调控提供了新的视角。