Novo Nordisk Foundation Center for Protein Research, Department of Health and Medical Sciences, University of Copenhagen, 2200 Copenhagen N, Denmark.
Department of Drug Design and Pharmacology, Faculty of Health and Medical Sciences, University of Copenhagen, c/o the Danish Cancer Society, Strandboulevarden 49, DK-2100 Copenhagen Ø, Denmark.
Sci Rep. 2017 Feb 20;7:42800. doi: 10.1038/srep42800.
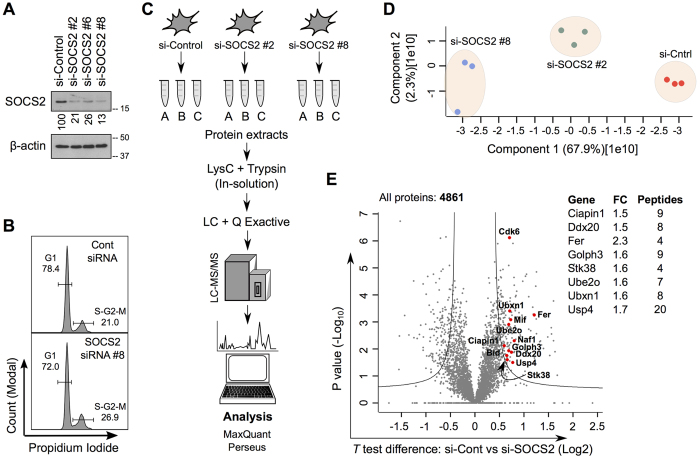
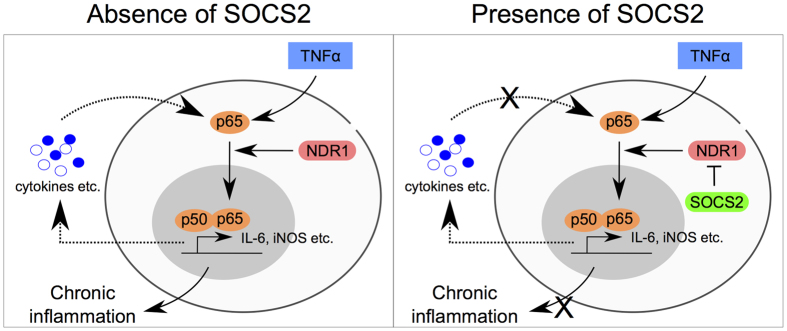
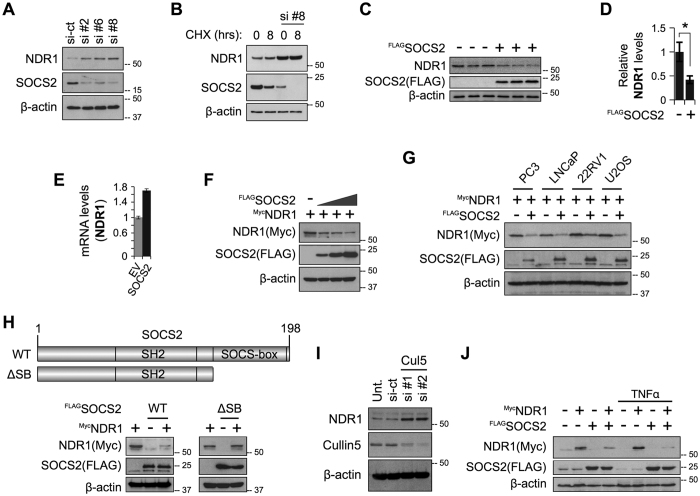
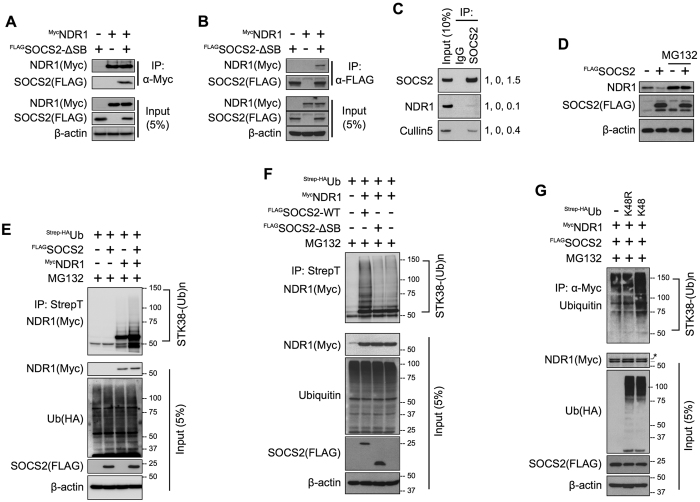
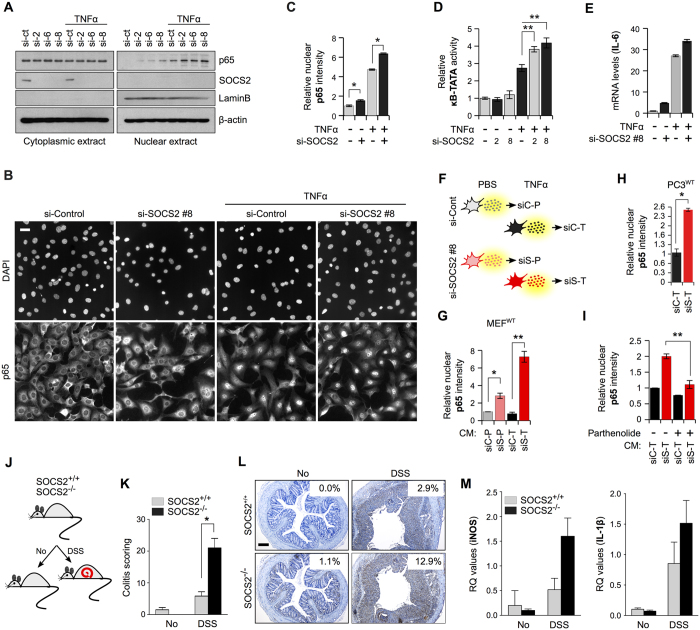
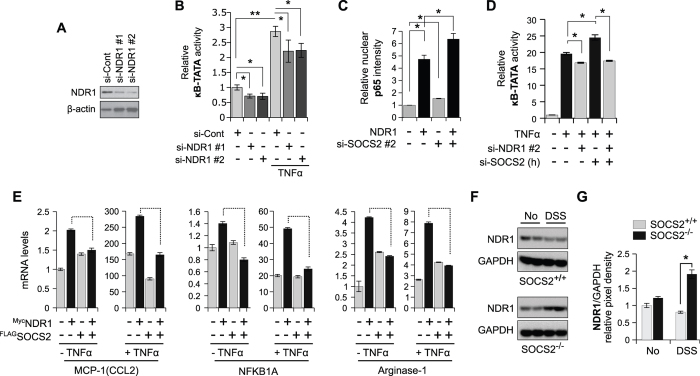
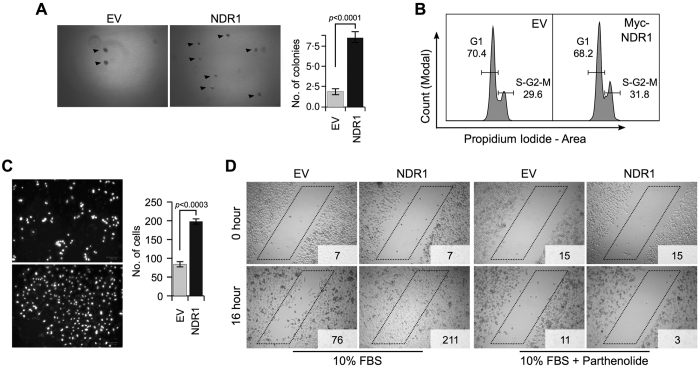
SOCS2 is a pleiotropic E3 ligase. Its deficiency is associated with gigantism and organismal lethality upon inflammatory challenge. However, mechanistic understanding of SOCS2 function is dismal due to our unawareness of its protein substrates. We performed a mass spectrometry based proteomic profiling upon SOCS2 depletion and yield quantitative data for ~4200 proteins. Through this screen we identify a novel target of SOCS2, the serine-threonine kinase NDR1. Over-expression of SOCS2 accelerates turnover, while its knockdown stabilizes, endogenous NDR1 protein. SOCS2 interacts with NDR1 and promotes its degradation through K48-linked ubiquitination. Functionally, over-expression of SOCS2 antagonizes NDR1-induced TNFα-stimulated NF-κB activity. Conversely, depletion of NDR1 rescues the effect of SOCS2-deficiency on TNFα-induced NF-κB transactivation. Using a SOCS2 mice model of colitis we show that SOCS2-deficiency is pro-inflammatory and negatively correlates with NDR1 and nuclear p65 levels. Lastly, we provide evidence to suggest that NDR1 acts as an oncogene in prostate cancer. To the best of our knowledge, this is the first report of an identified E3 ligase for NDR1. These results might explain how SOCS2-deficiency leads to hyper-activation of NF-κB and downstream pathological implications and posits that SOCS2 induced degradation of NDR1 may act as a switch in restricting TNFα-NF-κB pathway.
SOCS2 是一种多效性的 E3 连接酶。其缺乏与巨人症和炎症挑战时的机体致死性有关。然而,由于我们不知道 SOCS2 的蛋白质底物,因此对 SOCS2 功能的机制理解仍然很困难。我们在 SOCS2 耗尽后进行了基于质谱的蛋白质组学分析,获得了约 4200 种蛋白质的定量数据。通过这个筛选,我们确定了 SOCS2 的一个新靶标,丝氨酸-苏氨酸激酶 NDR1。SOCS2 的过表达加速了 NDR1 的周转率,而其敲低则稳定了内源性 NDR1 蛋白。SOCS2 与 NDR1 相互作用,并通过 K48 连接的泛素化促进其降解。功能上,SOCS2 的过表达拮抗了 NDR1 诱导的 TNFα 刺激的 NF-κB 活性。相反,NDR1 的耗竭挽救了 SOCS2 缺乏对 TNFα 诱导的 NF-κB 反式激活的影响。使用 SOCS2 结肠炎小鼠模型,我们表明 SOCS2 缺乏是促炎的,与 NDR1 和核 p65 水平呈负相关。最后,我们提供了证据表明 NDR1 是前列腺癌中的癌基因。据我们所知,这是首次报道鉴定出 NDR1 的 E3 连接酶。这些结果可能解释了 SOCS2 缺乏如何导致 NF-κB 的过度激活以及下游的病理影响,并假设 SOCS2 诱导的 NDR1 降解可能作为限制 TNFα-NF-κB 途径的开关。