Stirnemann Jérôme, Belmatoug Nadia, Camou Fabrice, Serratrice Christine, Froissart Roseline, Caillaud Catherine, Levade Thierry, Astudillo Leonardo, Serratrice Jacques, Brassier Anaïs, Rose Christian, Billette de Villemeur Thierry, Berger Marc G
Department of Internal Medicine, Geneva University Hospital, Rue Gabrielle-Perret-Gentil 4, CH-1211 Genève, Switzerland.
Department of Internal Medicine, Reference Center for Lysosomal Storage Diseases, Hôpitaux Universitaires Paris Nord Val de Seine, site Beaujon, Assistance Publique-Hôpitaux de Paris, 100 boulevard du Général Leclerc, F-92110 Clichy la Garenne, France.
Int J Mol Sci. 2017 Feb 17;18(2):441. doi: 10.3390/ijms18020441.
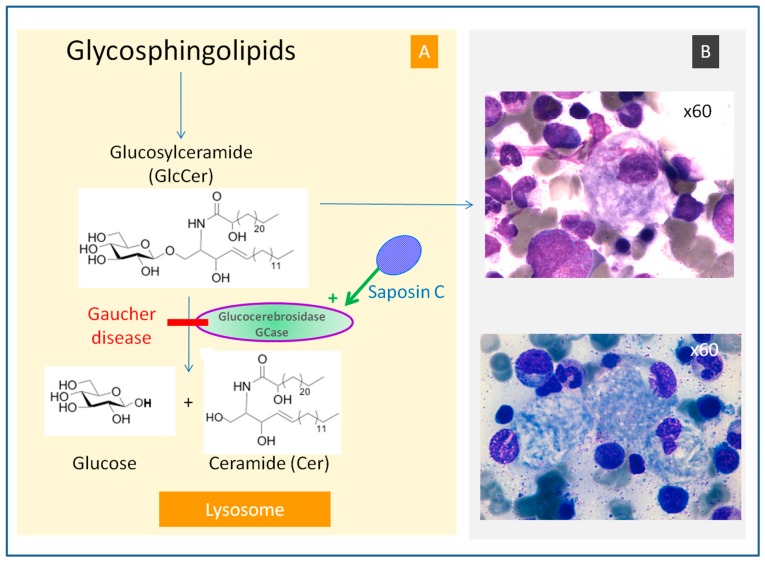
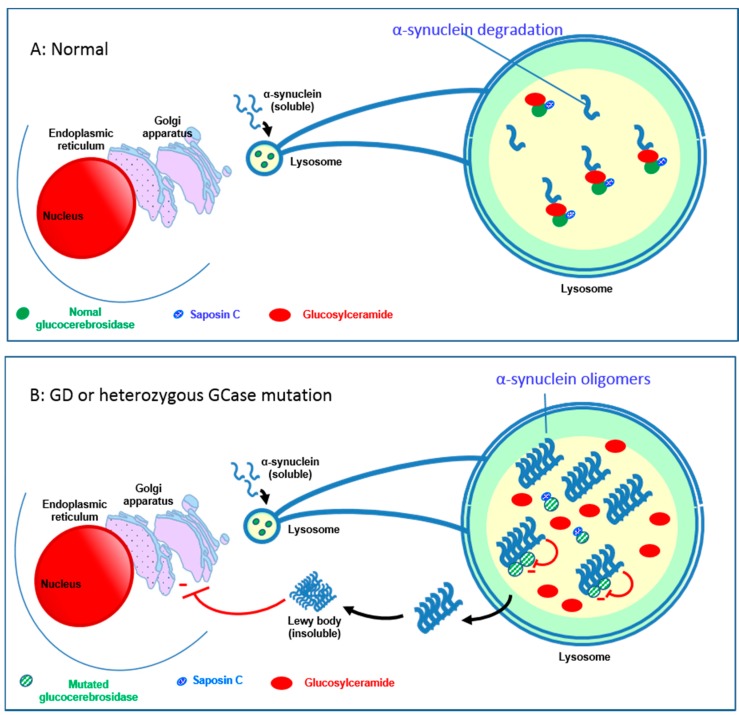
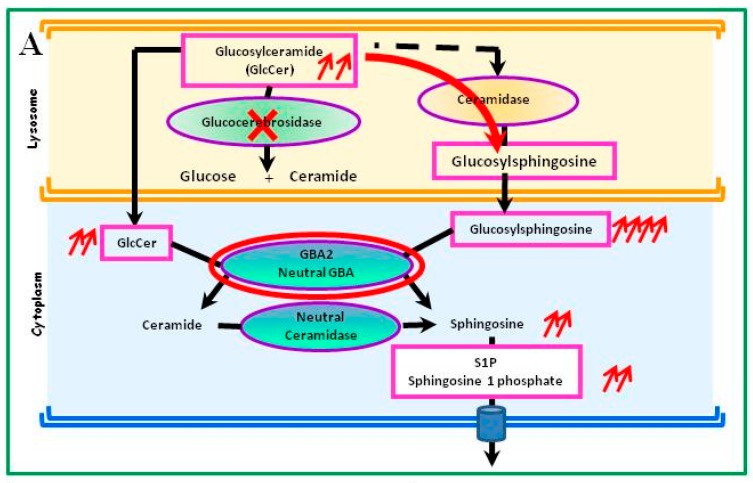
Gaucher disease (GD, ORPHA355) is a rare, autosomal recessive genetic disorder. It is caused by a deficiency of the lysosomal enzyme, glucocerebrosidase, which leads to an accumulation of its substrate, glucosylceramide, in macrophages. In the general population, its incidence is approximately 1/40,000 to 1/60,000 births, rising to 1/800 in Ashkenazi Jews. The main cause of the cytopenia, splenomegaly, hepatomegaly, and bone lesions associated with the disease is considered to be the infiltration of the bone marrow, spleen, and liver by Gaucher cells. Type-1 Gaucher disease, which affects the majority of patients (90% in Europe and USA, but less in other regions), is characterized by effects on the viscera, whereas types 2 and 3 are also associated with neurological impairment, either severe in type 2 or variable in type 3. A diagnosis of GD can be confirmed by demonstrating the deficiency of acid glucocerebrosidase activity in leukocytes. Mutations in the gene should be identified as they may be of prognostic value in some cases. Patients with type-1 GD-but also carriers of mutation-have been found to be predisposed to developing Parkinson's disease, and the risk of neoplasia associated with the disease is still subject to discussion. Disease-specific treatment consists of intravenous enzyme replacement therapy (ERT) using one of the currently available molecules (imiglucerase, velaglucerase, or taliglucerase). Orally administered inhibitors of glucosylceramide biosynthesis can also be used (miglustat or eliglustat).
戈谢病(GD,孤儿病编号355)是一种罕见的常染色体隐性遗传病。它由溶酶体酶葡萄糖脑苷脂酶缺乏引起,导致其底物葡萄糖神经酰胺在巨噬细胞中蓄积。在普通人群中,其发病率约为每40000至60000例出生中有1例,在阿什肯纳兹犹太人中升至每800例出生中有1例。与该疾病相关的血细胞减少、脾肿大、肝肿大和骨病变的主要原因被认为是戈谢细胞浸润骨髓、脾脏和肝脏。1型戈谢病影响大多数患者(在欧洲和美国为90%,但在其他地区比例较低),其特征是对内脏产生影响,而2型和3型还与神经功能障碍有关,2型严重,3型程度不一。通过证明白细胞中酸性葡萄糖脑苷脂酶活性缺乏可确诊戈谢病。应鉴定该基因的突变,因为在某些情况下它们可能具有预后价值。已发现1型戈谢病患者以及该基因突变携带者易患帕金森病,与该疾病相关的肿瘤形成风险仍在讨论中。针对该疾病的治疗包括使用目前可用的一种分子(伊米苷酶、维拉苷酶或他利苷酶)进行静脉酶替代疗法(ERT)。也可使用口服的葡萄糖神经酰胺生物合成抑制剂(米格列醇或依利格鲁司他)。