Romano Valentina, de Beer Tjaart A P, Schwede Torsten
Biozentrum, University of Basel, Basel, Switzerland.
SIB Swiss Institute of Bioinformatics, Basel, Switzerland.
BMC Res Notes. 2017 Feb 20;10(1):104. doi: 10.1186/s13104-017-2428-9.
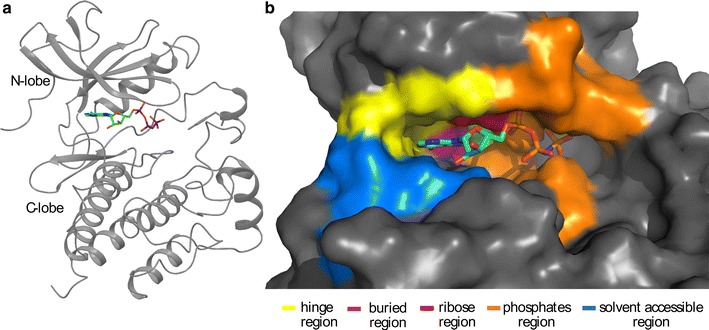
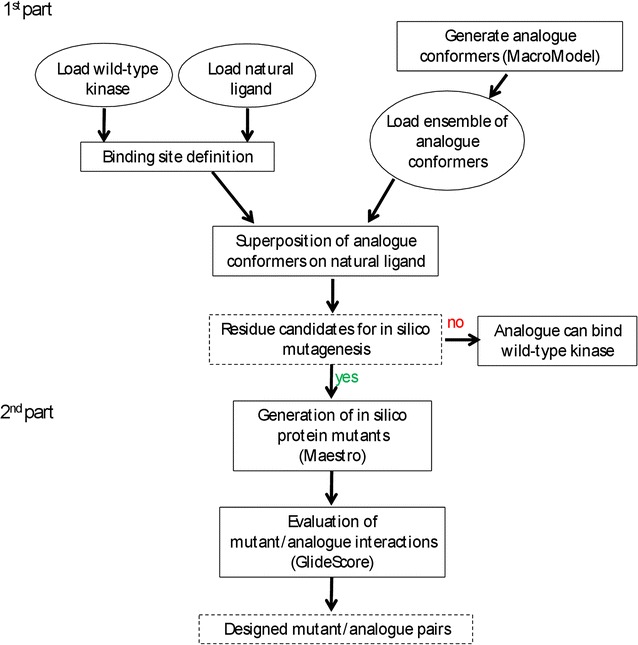
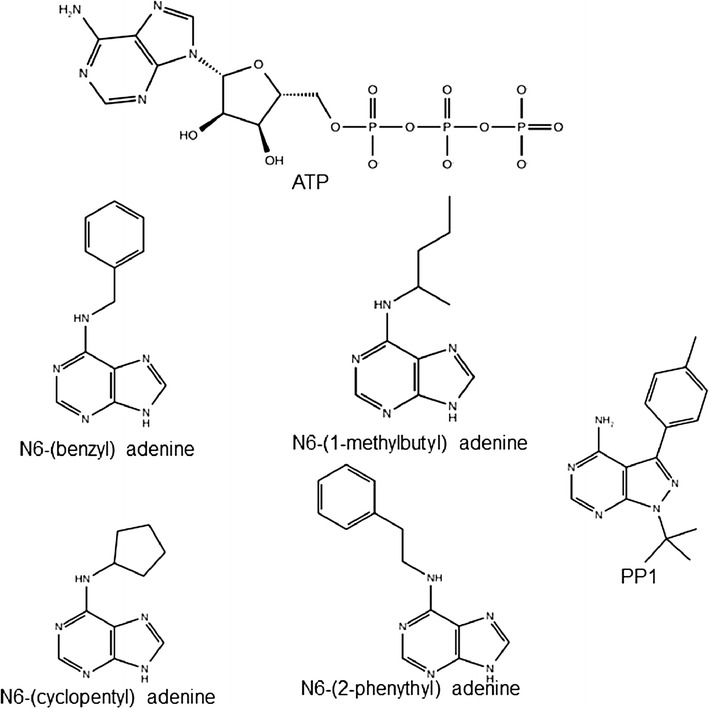
The determination of specific kinase substrates in vivo is challenging due to the large number of protein kinases in cells, their substrate specificity overlap, and the lack of highly specific inhibitors. In the late 90s, Shokat and coworkers developed a protein engineering-based method addressing the question of identification of substrates of protein kinases. The approach was based on the mutagenesis of the gatekeeper residue within the binding site of a protein kinase to change the co-substrate specificity from ATP to ATP analogues. One of the challenges in applying this method to other kinase systems is to identify the optimal combination of mutation in the enzyme and chemical derivative such that the ATP analogue acts as substrate for the engineered, but not the native kinase enzyme. In this study, we developed a computational protocol for estimating the effect of mutations at the gatekeeper position on the accessibility of ATP analogues within the binding site of engineered kinases.
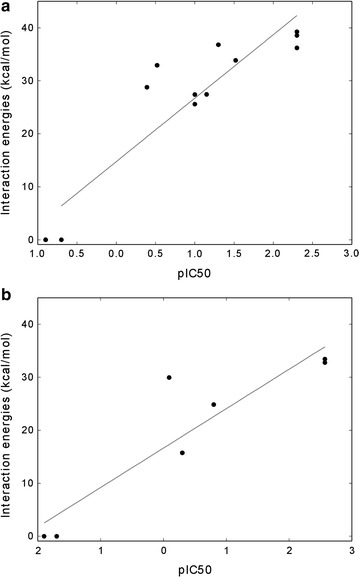
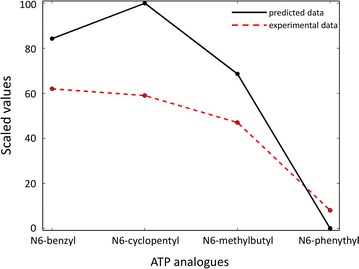
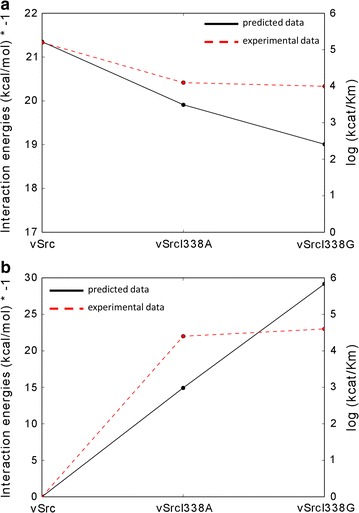
We tested the protocol on a dataset of tyrosine and serine/threonine protein kinases from the scientific literature where Shokat's method was applied and experimental data were available. Our protocol correctly identified gatekeeper residues as the positions to mutate within the binding site of the studied kinase enzymes. Furthermore, the approach well reproduced the experimental data available in literature.
We have presented a computational protocol that scores how different mutations at the gatekeeper position influence the accommodation of various ATP analogues within the binding site of protein kinases. We have assessed our approach on protein kinases from the scientific literature and have verified the ability of the approach to well reproduce the available experimental data and identify suitable combinations of engineered kinases and ATP analogues.
由于细胞中存在大量蛋白激酶,它们的底物特异性存在重叠,且缺乏高度特异性抑制剂,因此在体内确定特定激酶底物具有挑战性。在20世纪90年代后期,肖卡特及其同事开发了一种基于蛋白质工程的方法来解决蛋白激酶底物鉴定问题。该方法基于对蛋白激酶结合位点内的守门残基进行诱变,以将共底物特异性从ATP改变为ATP类似物。将该方法应用于其他激酶系统的挑战之一是确定酶中的突变与化学衍生物的最佳组合,以使ATP类似物作为工程化激酶而非天然激酶的底物。在本研究中,我们开发了一种计算方案,用于估计守门位置的突变对工程化激酶结合位点内ATP类似物可及性的影响。
我们在来自科学文献的酪氨酸和丝氨酸/苏氨酸蛋白激酶数据集上测试了该方案,这些文献应用了肖卡特的方法且有实验数据。我们的方案正确地将守门残基识别为所研究激酶酶结合位点内要突变 的位置。此外,该方法很好地重现了文献中的实验数据。
我们提出了一种计算方案,该方案对守门位置的不同突变如何影响蛋白激酶结合位点内各种ATP类似物的容纳进行评分。我们已在来自科学文献的蛋白激酶上评估了我们的方法,并验证了该方法重现现有实验数据以及识别工程化激酶和ATP类似物合适组合的能力。