Behavioral Health Epidemiology Program, RTI International, 3040 Cornwallis Road, PO Box 12194, Research Triangle Park, NC 27709-12194, USA.
Hum Genet. 2013 May;132(5):509-22. doi: 10.1007/s00439-013-1266-7. Epub 2013 Jan 22.
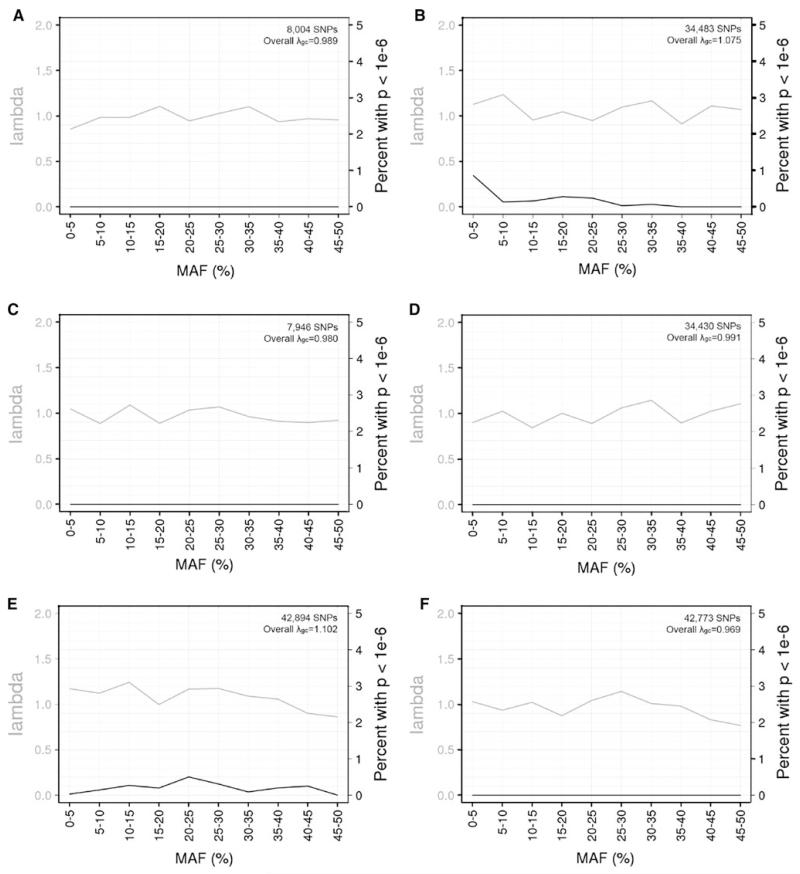
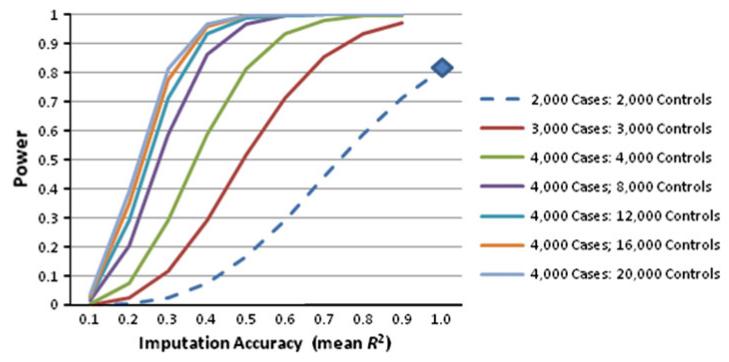
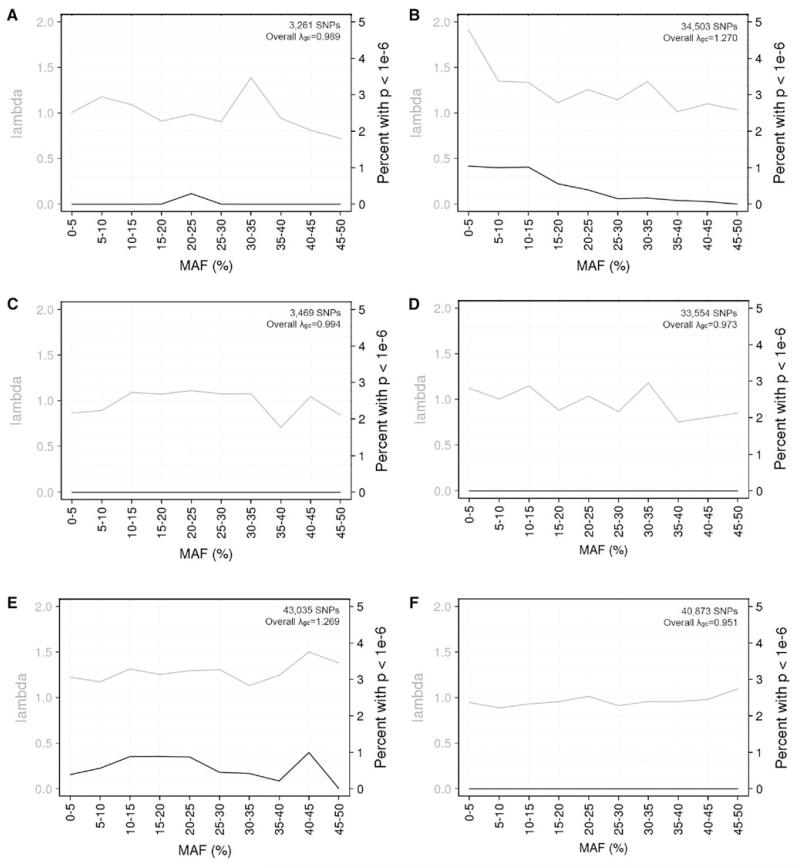
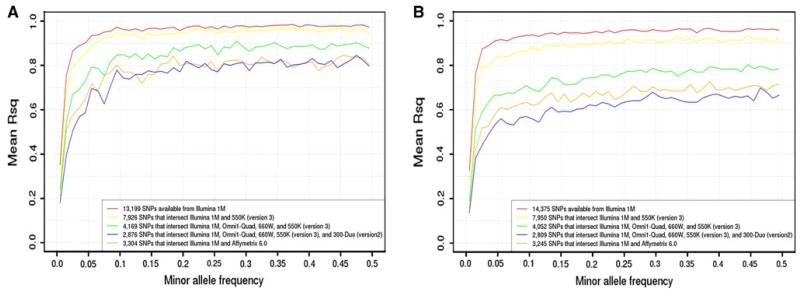
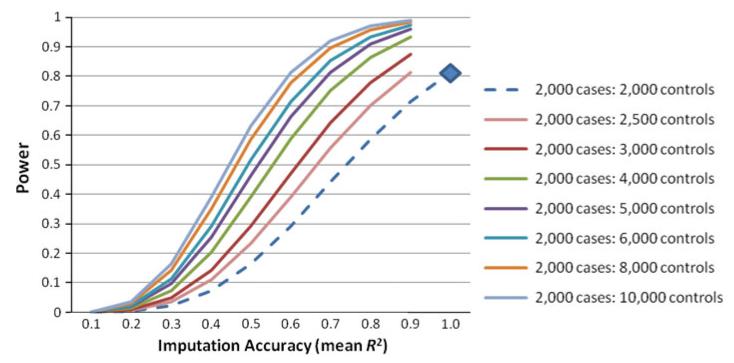
A great promise of publicly sharing genome-wide association data is the potential to create composite sets of controls. However, studies often use different genotyping arrays, and imputation to a common set of SNPs has shown substantial bias: a problem which has no broadly applicable solution. Based on the idea that using differing genotyped SNP sets as inputs creates differential imputation errors and thus bias in the composite set of controls, we examined the degree to which each of the following occurs: (1) imputation based on the union of genotyped SNPs (i.e., SNPs available on one or more arrays) results in bias, as evidenced by spurious associations (type 1 error) between imputed genotypes and arbitrarily assigned case/control status; (2) imputation based on the intersection of genotyped SNPs (i.e., SNPs available on all arrays) does not evidence such bias; and (3) imputation quality varies by the size of the intersection of genotyped SNP sets. Imputations were conducted in European Americans and African Americans with reference to HapMap phase II and III data. Imputation based on the union of genotyped SNPs across the Illumina 1M and 550v3 arrays showed spurious associations for 0.2 % of SNPs: ~2,000 false positives per million SNPs imputed. Biases remained problematic for very similar arrays (550v1 vs. 550v3) and were substantial for dissimilar arrays (Illumina 1M vs. Affymetrix 6.0). In all instances, imputing based on the intersection of genotyped SNPs (as few as 30 % of the total SNPs genotyped) eliminated such bias while still achieving good imputation quality.
公开分享全基因组关联数据的一个巨大承诺是有可能创建复合对照组。然而,研究通常使用不同的基因分型阵列,而对共同的 SNP 集进行推断表明存在大量偏差:这是一个没有广泛适用解决方案的问题。基于使用不同的基因分型 SNP 集作为输入会产生不同的推断错误,从而导致对照组的偏差的想法,我们检查了以下每种情况发生的程度:(1)基于基因分型 SNP 的并集(即,一个或多个阵列上可用的 SNP)进行推断会导致偏差,表现为推断基因型与任意分配的病例/对照组状态之间的虚假关联(类型 1 错误);(2)基于基因分型 SNP 的交集(即,所有阵列上可用的 SNP)进行推断不会出现这种偏差;(3)基因分型 SNP 集的交集大小会影响推断质量。在欧洲裔美国人和非裔美国人中进行了推断,参考 HapMap 第二阶段和第三阶段的数据。基于 Illumina 1M 和 550v3 阵列上基因分型 SNP 的并集进行推断会导致 0.2%的 SNP 出现虚假关联:每百万 SNP 推断中约有 2000 个假阳性。非常相似的阵列(550v1 与 550v3)之间的偏差仍然存在问题,而不相似的阵列(Illumina 1M 与 Affymetrix 6.0)之间的偏差则更为严重。在所有情况下,基于基因分型 SNP 的交集(即使只有总 SNP 中 30%的 SNP 进行了基因分型)进行推断可以消除这种偏差,同时仍然实现良好的推断质量。