De Maio Nicola, Wilson Daniel J
Institute for Emerging Infections, Oxford Martin School, University of Oxford, Oxford, OX1 3PA, United Kingdom
Nuffield Department of Medicine, University of Oxford, Oxford, OX1 3PA, United Kingdom.
Genetics. 2017 May;206(1):333-343. doi: 10.1534/genetics.116.198796. Epub 2017 Mar 3.
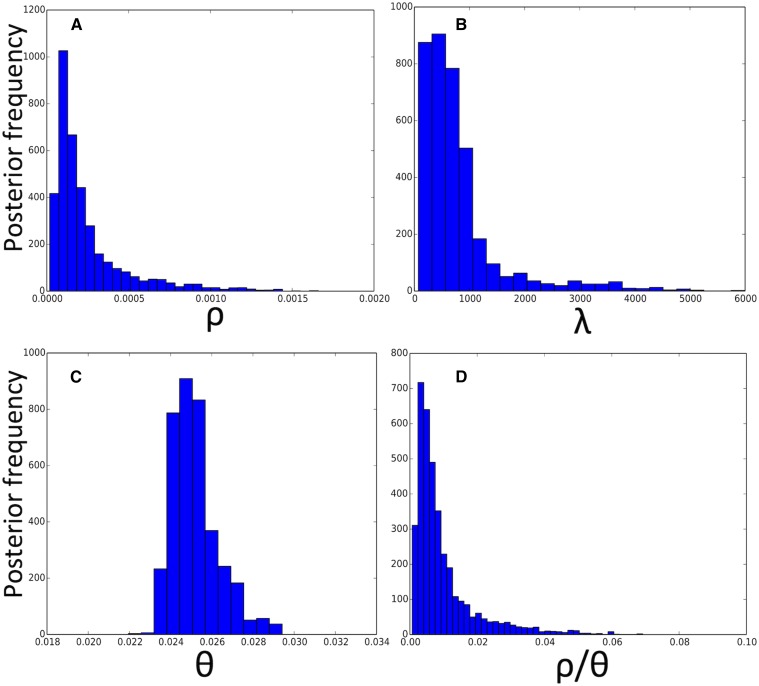
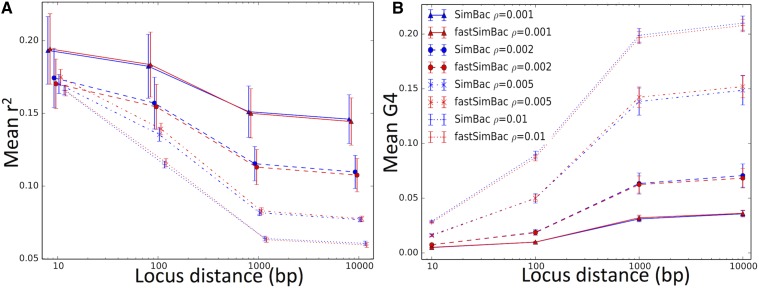
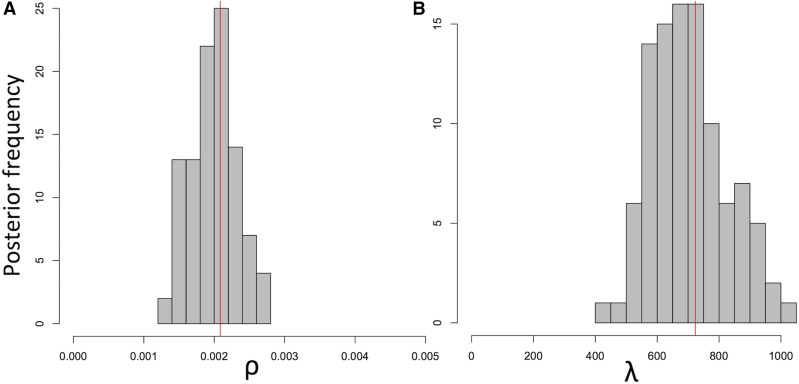
Bacteria can exchange and acquire new genetic material from other organisms directly and via the environment. This process, known as bacterial recombination, has a strong impact on the evolution of bacteria, for example, leading to the spread of antibiotic resistance across clades and species, and to the avoidance of clonal interference. Recombination hinders phylogenetic and transmission inference because it creates patterns of substitutions (homoplasies) inconsistent with the hypothesis of a single evolutionary tree. Bacterial recombination is typically modeled as statistically akin to gene conversion in eukaryotes, , using the coalescent with gene conversion (CGC). However, this model can be very computationally demanding as it needs to account for the correlations of evolutionary histories of even distant loci. So, with the increasing popularity of whole genome sequencing, the need has emerged for a faster approach to model and simulate bacterial genome evolution. We present a new model that approximates the coalescent with gene conversion: the bacterial sequential Markov coalescent (BSMC). Our approach is based on a similar idea to the sequential Markov coalescent (SMC)-an approximation of the coalescent with crossover recombination. However, bacterial recombination poses hurdles to a sequential Markov approximation, as it leads to strong correlations and linkage disequilibrium across very distant sites in the genome. Our BSMC overcomes these difficulties, and shows a considerable reduction in computational demand compared to the exact CGC, and very similar patterns in simulated data. We implemented our BSMC model within new simulation software FastSimBac. In addition to the decreased computational demand compared to previous bacterial genome evolution simulators, FastSimBac provides more general options for evolutionary scenarios, allowing population structure with migration, speciation, population size changes, and recombination hotspots. FastSimBac is available from https://bitbucket.org/nicofmay/fastsimbac, and is distributed as open source under the terms of the GNU General Public License. Lastly, we use the BSMC within an Approximate Bayesian Computation (ABC) inference scheme, and suggest that parameters simulated under the exact CGC can correctly be recovered, further showcasing the accuracy of the BSMC. With this ABC we infer recombination rate, mutation rate, and recombination tract length of from a whole genome alignment.
细菌可以直接或通过环境从其他生物体交换并获取新的遗传物质。这一过程被称为细菌重组,对细菌的进化有很大影响,例如,导致抗生素抗性在进化枝和物种间传播,并避免克隆干扰。重组会阻碍系统发育和传播推断,因为它会产生与单一进化树假设不一致的替代模式(同塑性)。细菌重组通常在统计上被建模为类似于真核生物中的基因转换,即使用带有基因转换的合并模型(CGC)。然而,该模型的计算量可能非常大,因为它需要考虑即使是距离很远的基因座的进化历史相关性。因此,随着全基因组测序越来越普及,出现了对一种更快的方法来建模和模拟细菌基因组进化的需求。我们提出了一种新的模型,即细菌序列马尔可夫合并模型(BSMC),它近似于带有基因转换的合并模型。我们的方法基于与序列马尔可夫合并模型(SMC)类似的理念——一种带有交叉重组的合并模型的近似。然而,细菌重组给序列马尔可夫近似带来了障碍,因为它会导致基因组中非常远的位点之间产生强相关性和连锁不平衡。我们的BSMC克服了这些困难,与精确的CGC相比,计算需求大幅降低,并且模拟数据中的模式非常相似。我们在新的模拟软件FastSimBac中实现了我们的BSMC模型。除了与之前的细菌基因组进化模拟器相比计算需求降低外,FastSimBac还为进化场景提供了更通用的选项,允许有迁移、物种形成、种群大小变化和重组热点的种群结构。FastSimBac可从https://bitbucket.org/nicofmay/fastsimbac获取,并根据GNU通用公共许可证的条款作为开源软件分发。最后,我们在近似贝叶斯计算(ABC)推理方案中使用了BSMC,并表明在精确的CGC下模拟的参数可以被正确恢复,进一步展示了BSMC的准确性。通过这种ABC方法,我们从全基因组比对中推断出重组率、突变率和重组片段长度。