European Molecular Biology Laboratory, European Bioinformatics Institute, Hinxton, United Kingdom.
Department of Genetics, University of Cambridge, Cambridge, United Kingdom.
PLoS Comput Biol. 2022 Apr 29;18(4):e1010056. doi: 10.1371/journal.pcbi.1010056. eCollection 2022 Apr.
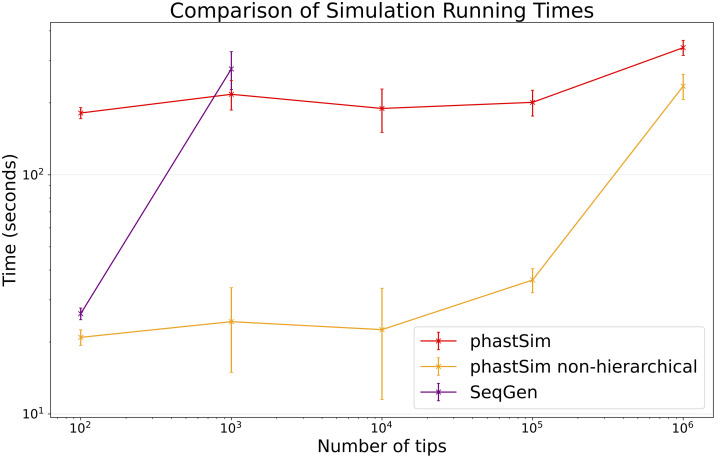
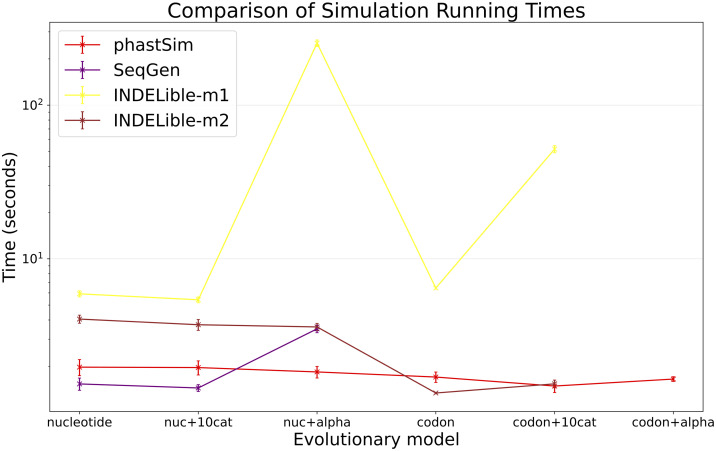
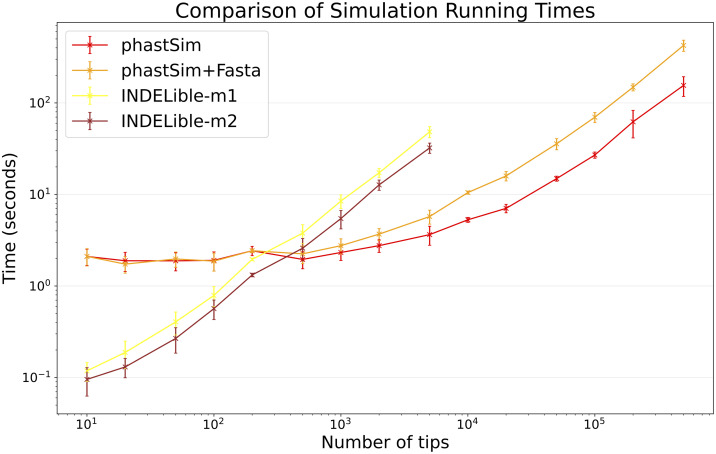
Sequence simulators are fundamental tools in bioinformatics, as they allow us to test data processing and inference tools, and are an essential component of some inference methods. The ongoing surge in available sequence data is however testing the limits of our bioinformatics software. One example is the large number of SARS-CoV-2 genomes available, which are beyond the processing power of many methods, and simulating such large datasets is also proving difficult. Here, we present a new algorithm and software for efficiently simulating sequence evolution along extremely large trees (e.g. > 100, 000 tips) when the branches of the tree are short, as is typical in genomic epidemiology. Our algorithm is based on the Gillespie approach, and it implements an efficient multi-layered search tree structure that provides high computational efficiency by taking advantage of the fact that only a small proportion of the genome is likely to mutate at each branch of the considered phylogeny. Our open source software allows easy integration with other Python packages as well as a variety of evolutionary models, including indel models and new hypermutability models that we developed to more realistically represent SARS-CoV-2 genome evolution.
序列模拟器是生物信息学中的基本工具,因为它们允许我们测试数据处理和推理工具,并且是一些推理方法的重要组成部分。然而,可用序列数据的持续激增正在考验我们生物信息学软件的极限。一个例子是大量的 SARS-CoV-2 基因组可用,这超出了许多方法的处理能力,并且模拟如此大规模的数据集也证明是困难的。在这里,我们提出了一种新的算法和软件,用于在树的分支较短时有效地模拟序列进化,例如 > 100,000 个叶(tips),这在基因组流行病学中是典型的。我们的算法基于 Gillespie 方法,并实现了一种有效的多层搜索树结构,通过利用只有一小部分基因组可能在考虑的系统发育的每个分支上发生突变的事实,提供了高效的计算效率。我们的开源软件允许与其他 Python 包以及各种进化模型(包括我们开发的插入缺失模型和新的高突变模型)轻松集成,以更真实地表示 SARS-CoV-2 基因组进化。