Melbourne Integrative Genomics / School of Mathematics and Statistics, The University of Melbourne, Melbourne, Australia.
Department of Statistics, The University of Warwick, Coventry, United Kingdom.
PLoS Comput Biol. 2022 Mar 9;18(3):e1009960. doi: 10.1371/journal.pcbi.1009960. eCollection 2022 Mar.
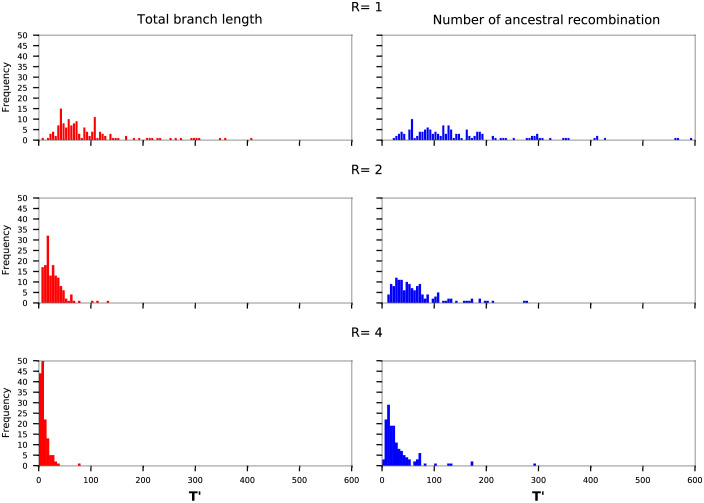
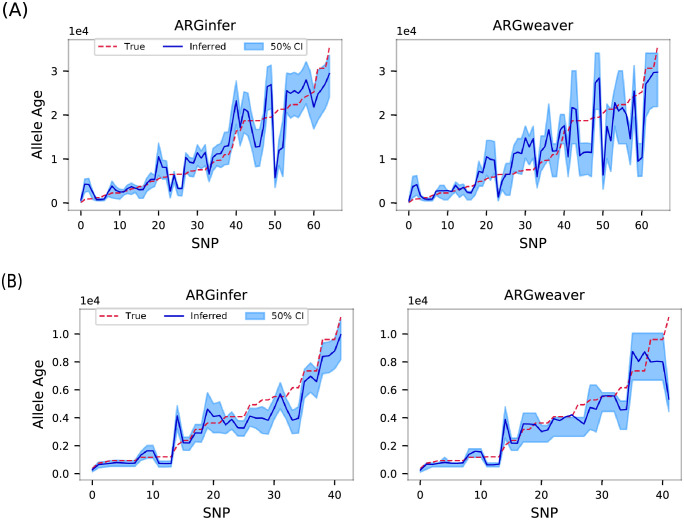
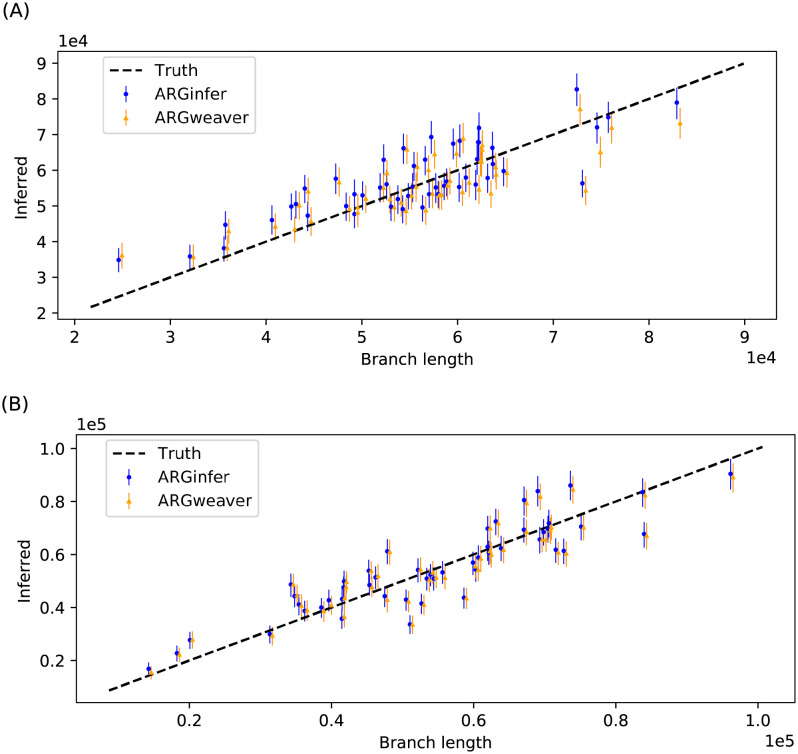
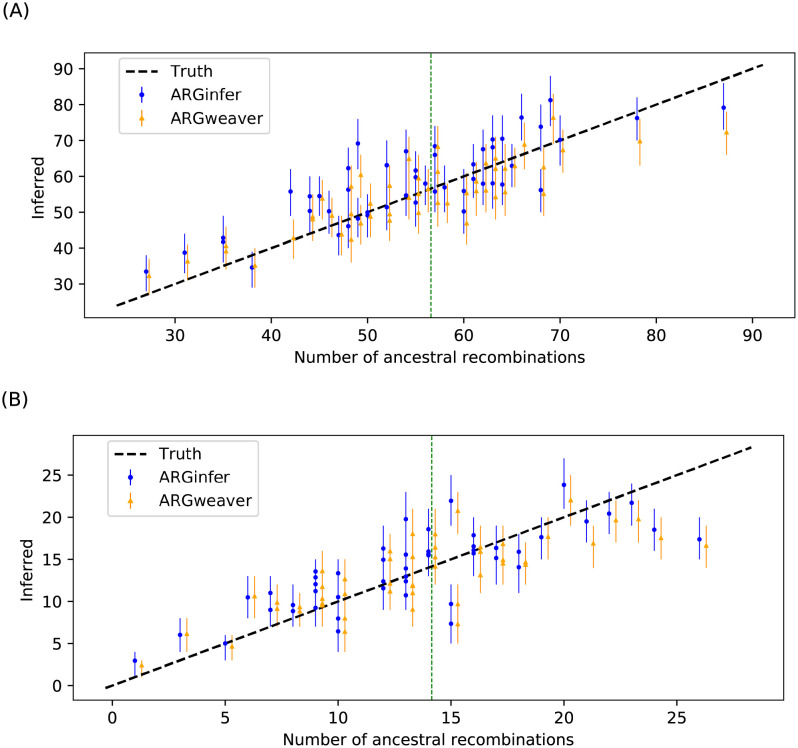
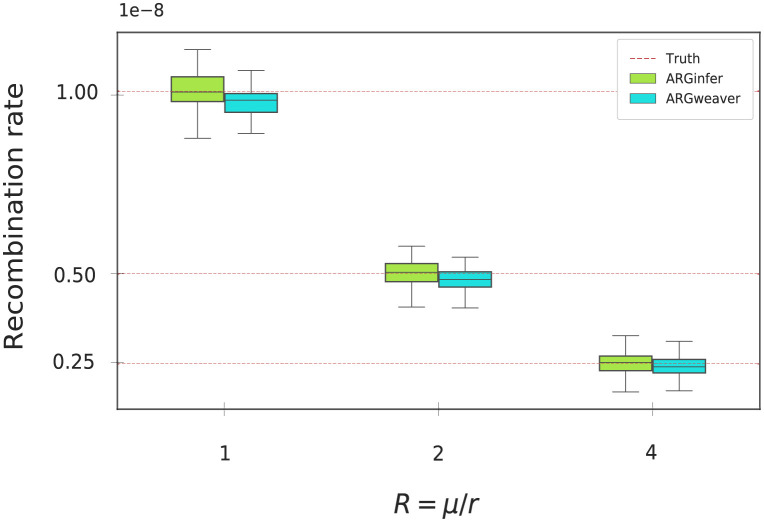
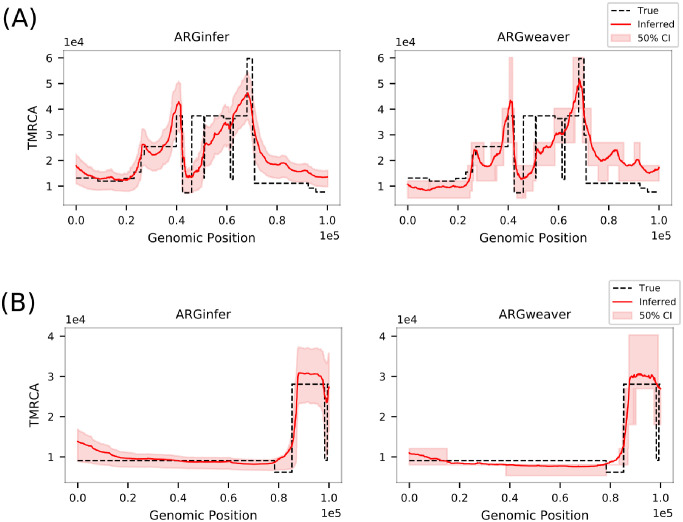
We present a novel algorithm, implemented in the software ARGinfer, for probabilistic inference of the Ancestral Recombination Graph under the Coalescent with Recombination. Our Markov Chain Monte Carlo algorithm takes advantage of the Succinct Tree Sequence data structure that has allowed great advances in simulation and point estimation, but not yet probabilistic inference. Unlike previous methods, which employ the Sequentially Markov Coalescent approximation, ARGinfer uses the Coalescent with Recombination, allowing more accurate inference of key evolutionary parameters. We show using simulations that ARGinfer can accurately estimate many properties of the evolutionary history of the sample, including the topology and branch lengths of the genealogical tree at each sequence site, and the times and locations of mutation and recombination events. ARGinfer approximates posterior probability distributions for these and other quantities, providing interpretable assessments of uncertainty that we show to be well calibrated. ARGinfer is currently limited to tens of DNA sequences of several hundreds of kilobases, but has scope for further computational improvements to increase its applicability.
我们提出了一种新的算法,在软件 ARGinfer 中实现,用于在带有重组的合并下对祖先重组图进行概率推断。我们的马尔可夫链蒙特卡罗算法利用了简洁树序列数据结构,这使得模拟和点估计取得了很大的进展,但尚未实现概率推断。与之前使用连续马尔可夫合并近似的方法不同,ARGinfer 使用带有重组的合并,从而可以更准确地推断关键进化参数。我们通过模拟表明,ARGinfer 可以准确估计样本进化历史的许多属性,包括每个序列位置的系统发育树的拓扑结构和分支长度,以及突变和重组事件的时间和位置。ARGinfer 对这些和其他数量的后验概率分布进行了近似,提供了可解释的不确定性评估,我们表明这些评估是经过良好校准的。ARGinfer 目前仅限于几十条数百千碱基的 DNA 序列,但有进一步提高计算效率的空间,以增加其适用性。