Physics Department, Universitat de les Illes Balears, Palma de Mallorca, Spain.
Sci Rep. 2017 Mar 6;7:43738. doi: 10.1038/srep43738.
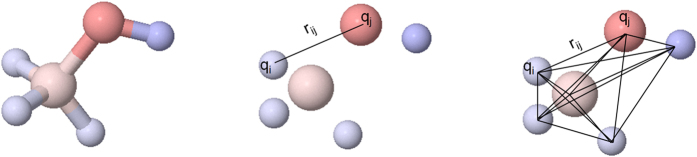
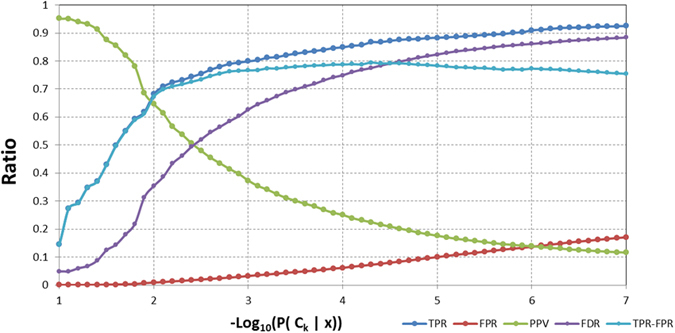
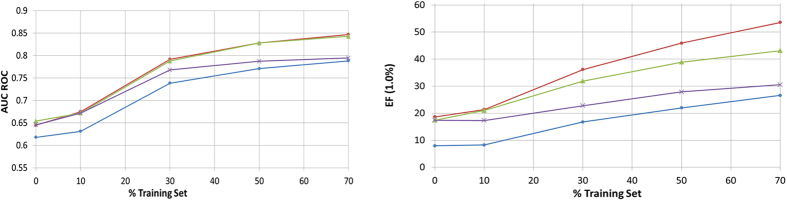
Virtual screening (VS) is applied in the early drug discovery phases for the quick inspection of huge molecular databases to identify those compounds that most likely bind to a given drug target. In this context, there is the necessity of the use of compact molecular models for database screening and precise target prediction in reasonable times. In this work we present a new compact energy-based model that is tested for its application to Virtual Screening and target prediction. The model can be used to quickly identify active compounds in huge databases based on the estimation of the molecule's pairing energies. The greatest molecular polar regions along with its geometrical distribution are considered by using a short set of smart energy vectors. The model is tested using similarity searches within the Directory of Useful Decoys (DUD) database. The results obtained are considerably better than previously published models. As a Target prediction methodology we propose the use of a Bayesian Classifier that uses a combination of different active compounds to build an energy-dependent probability distribution function for each target.
虚拟筛选 (VS) 在药物发现的早期阶段被应用,用于快速检查庞大的分子数据库,以识别最有可能与特定药物靶标结合的化合物。在这种情况下,有必要使用紧凑的分子模型来进行数据库筛选,并在合理的时间内进行精确的靶标预测。在这项工作中,我们提出了一种新的基于能量的紧凑模型,用于虚拟筛选和靶标预测的应用。该模型可用于根据分子对能的估计,快速识别庞大数据库中的活性化合物。通过使用一组智能能量向量来考虑最大的分子极性区域及其几何分布。该模型使用目录中的相似性搜索有用的诱饵 (DUD) 数据库进行测试。所得到的结果明显优于以前发表的模型。作为一种靶标预测方法,我们提出使用贝叶斯分类器,该分类器使用不同的活性化合物的组合,为每个靶标构建一个能量相关的概率分布函数。