Depienne Christel, Nava Caroline, Keren Boris, Heide Solveig, Rastetter Agnès, Passemard Sandrine, Chantot-Bastaraud Sandra, Moutard Marie-Laure, Agrawal Pankaj B, VanNoy Grace, Stoler Joan M, Amor David J, Billette de Villemeur Thierry, Doummar Diane, Alby Caroline, Cormier-Daire Valérie, Garel Catherine, Marzin Pauline, Scheidecker Sophie, de Saint-Martin Anne, Hirsch Edouard, Korff Christian, Bottani Armand, Faivre Laurence, Verloes Alain, Orzechowski Christine, Burglen Lydie, Leheup Bruno, Roume Joelle, Andrieux Joris, Sheth Frenny, Datar Chaitanya, Parker Michael J, Pasquier Laurent, Odent Sylvie, Naudion Sophie, Delrue Marie-Ange, Le Caignec Cédric, Vincent Marie, Isidor Bertrand, Renaldo Florence, Stewart Fiona, Toutain Annick, Koehler Udo, Häckl Birgit, von Stülpnagel Celina, Kluger Gerhard, Møller Rikke S, Pal Deb, Jonson Tord, Soller Maria, Verbeek Nienke E, van Haelst Mieke M, de Kovel Carolien, Koeleman Bobby, Monroe Glen, van Haaften Gijs, Attié-Bitach Tania, Boutaud Lucile, Héron Delphine, Mignot Cyril
IGBMC, CNRS UMR 7104/INSERM U964/Université de Strasbourg, 67400, Illkirch, France.
Laboratoires de Génétique, Institut de Génétique médicale d'Alsace, Hôpitaux Universitaires de Strasbourg, 67000, Strasbourg, France.
Hum Genet. 2017 Apr;136(4):463-479. doi: 10.1007/s00439-017-1772-0. Epub 2017 Mar 10.
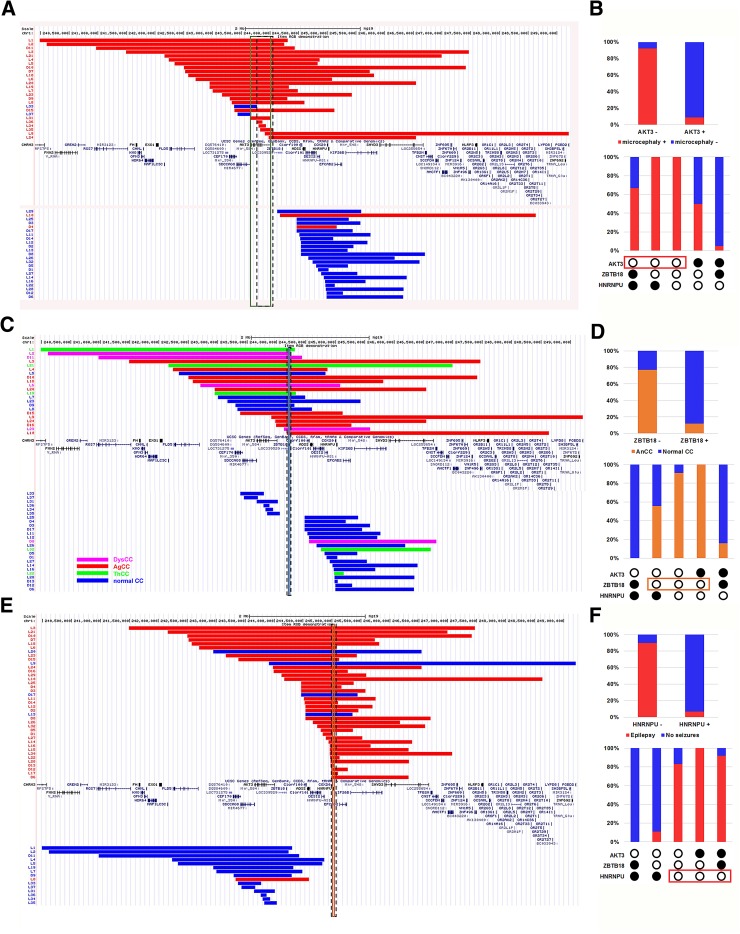
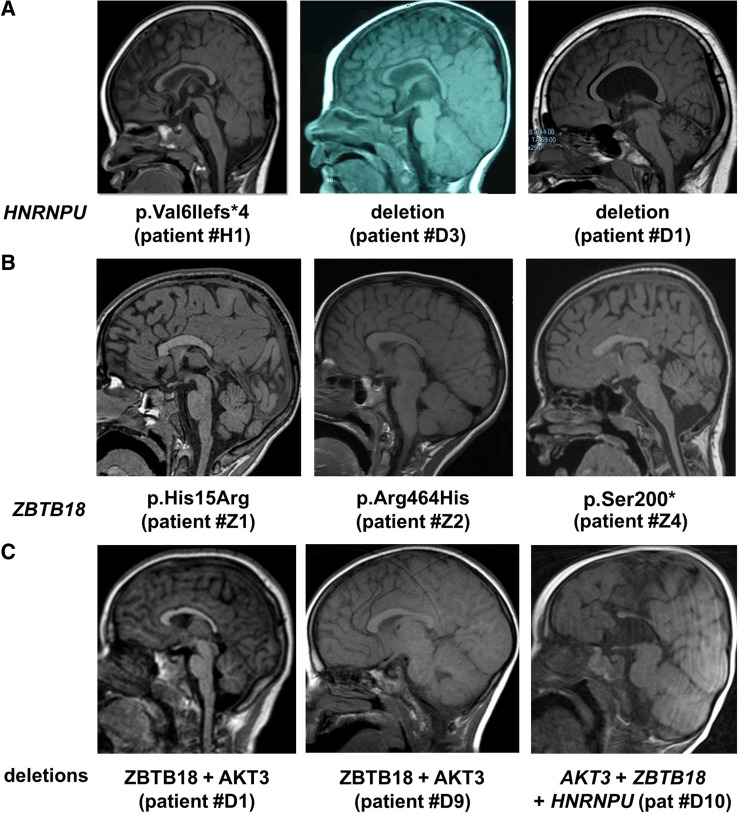
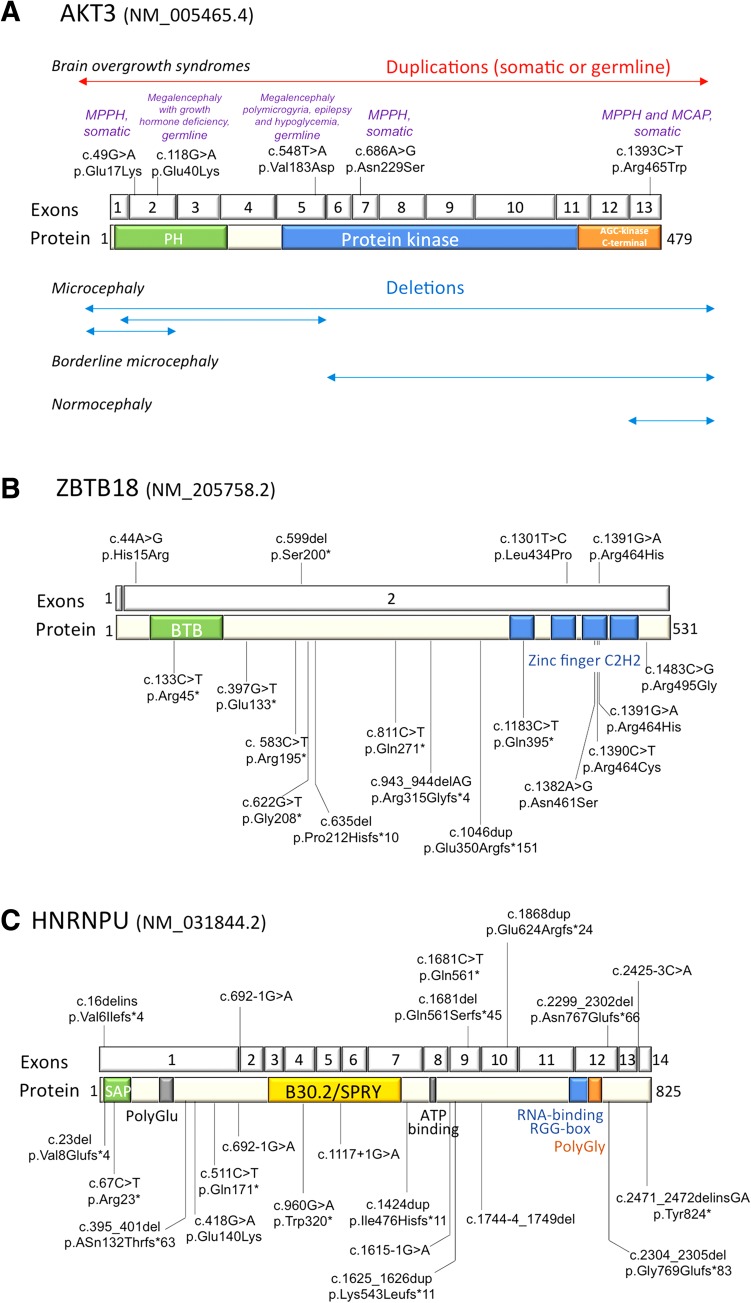
Subtelomeric 1q43q44 microdeletions cause a syndrome associating intellectual disability, microcephaly, seizures and anomalies of the corpus callosum. Despite several previous studies assessing genotype-phenotype correlations, the contribution of genes located in this region to the specific features of this syndrome remains uncertain. Among those, three genes, AKT3, HNRNPU and ZBTB18 are highly expressed in the brain and point mutations in these genes have been recently identified in children with neurodevelopmental phenotypes. In this study, we report the clinical and molecular data from 17 patients with 1q43q44 microdeletions, four with ZBTB18 mutations and seven with HNRNPU mutations, and review additional data from 37 previously published patients with 1q43q44 microdeletions. We compare clinical data of patients with 1q43q44 microdeletions with those of patients with point mutations in HNRNPU and ZBTB18 to assess the contribution of each gene as well as the possibility of epistasis between genes. Our study demonstrates that AKT3 haploinsufficiency is the main driver for microcephaly, whereas HNRNPU alteration mostly drives epilepsy and determines the degree of intellectual disability. ZBTB18 deletions or mutations are associated with variable corpus callosum anomalies with an incomplete penetrance. ZBTB18 may also contribute to microcephaly and HNRNPU to thin corpus callosum, but with a lower penetrance. Co-deletion of contiguous genes has additive effects. Our results confirm and refine the complex genotype-phenotype correlations existing in the 1qter microdeletion syndrome and define more precisely the neurodevelopmental phenotypes associated with genetic alterations of AKT3, ZBTB18 and HNRNPU in humans.
端粒亚端粒1q43q44微缺失导致一种综合征,伴有智力残疾、小头畸形、癫痫和胼胝体异常。尽管此前有多项研究评估了基因型与表型的相关性,但该区域基因对该综合征特定特征的贡献仍不明确。其中,AKT3、HNRNPU和ZBTB18这三个基因在大脑中高度表达,最近在患有神经发育表型的儿童中发现了这些基因的点突变。在本研究中,我们报告了17例1q43q44微缺失患者、4例ZBTB18突变患者和7例HNRNPU突变患者的临床和分子数据,并回顾了37例先前发表的1q43q44微缺失患者的其他数据。我们将1q43q44微缺失患者的临床数据与HNRNPU和ZBTB18点突变患者的临床数据进行比较,以评估每个基因的贡献以及基因之间上位性的可能性。我们的研究表明,AKT3单倍体不足是小头畸形的主要驱动因素,而HNRNPU改变主要驱动癫痫并决定智力残疾的程度。ZBTB18缺失或突变与胼胝体可变异常相关,且外显率不完全。ZBTB18也可能导致小头畸形,HNRNPU导致胼胝体变薄,但外显率较低。相邻基因的共同缺失具有累加效应。我们的结果证实并完善了1qter微缺失综合征中存在的复杂基因型-表型相关性,并更精确地定义了与人类AKT3、ZBTB18和HNRNPU基因改变相关的神经发育表型。