Zhu Lisha, Jiang Kaiyu, Webber Karstin, Wong Laiping, Liu Tao, Chen Yanmin, Jarvis James N
Department of Biochemistry, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo, NY, USA.
Department of Pediatrics, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo, NY, USA.
Arthritis Res Ther. 2017 Mar 14;19(1):57. doi: 10.1186/s13075-017-1260-x.
The transcriptomes of peripheral blood cells in children with juvenile idiopathic arthritis (JIA) have distinct transcriptional aberrations that suggest impairment of transcriptional regulation. To gain a better understanding of this phenomenon, we studied known JIA genetic risk loci, the majority of which are located in non-coding regions, where transcription is regulated and coordinated on a genome-wide basis. We examined human neutrophils and CD4 primary T cells to identify genes and functional elements located within those risk loci.
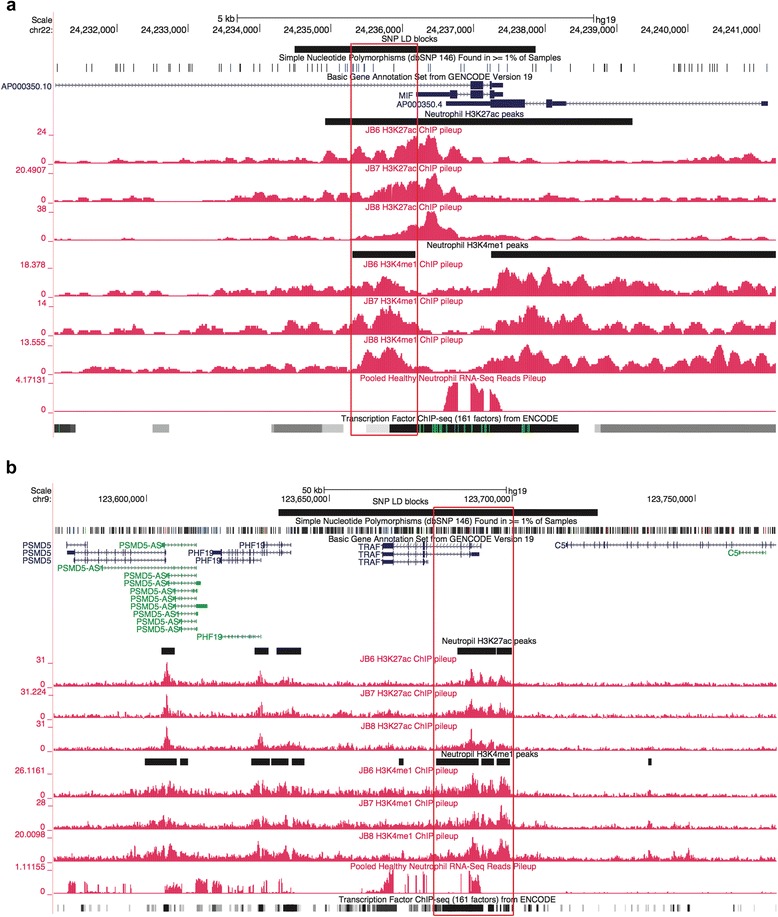
We analyzed RNA sequencing (RNA-Seq) data, H3K27ac and H3K4me1 chromatin immunoprecipitation-sequencing (ChIP-Seq) data, and previously published chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) data to characterize the chromatin landscapes within the known JIA-associated risk loci.
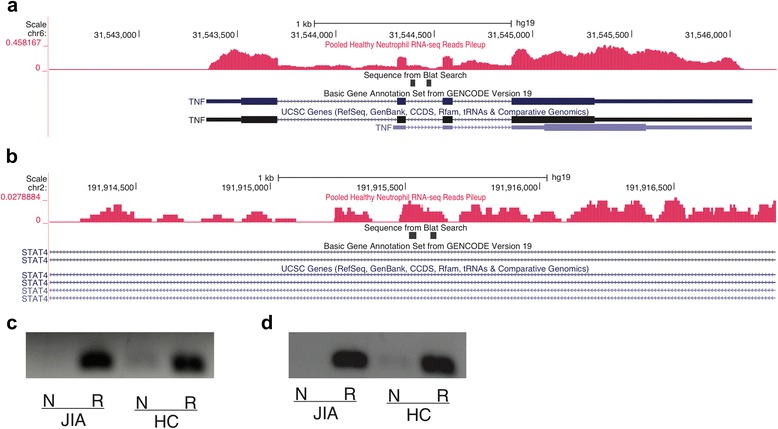
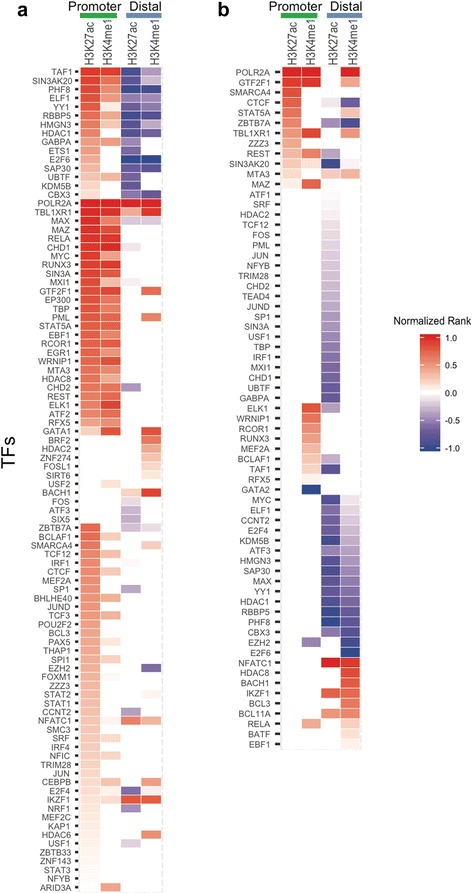
In both neutrophils and primary CD4+ T cells, the majority of the JIA-associated linkage disequilibrium (LD) blocks contained H3K27ac and/or H3K4me1 marks. These LD blocks were also binding sites for a small group of transcription factors, particularly in neutrophils. Furthermore, these regions showed abundant intronic and intergenic transcription in neutrophils. In neutrophils, none of the genes that were differentially expressed between untreated patients with JIA and healthy children were located within the JIA-risk LD blocks. In CD4+ T cells, multiple genes, including HLA-DQA1, HLA-DQB2, TRAF1, and IRF1 were associated with the long-distance interacting regions within the LD regions as determined from ChIA-PET data.
These findings suggest that genetic risk contributes to the aberrant transcriptional control observed in JIA. Furthermore, these findings demonstrate the challenges of identifying the actual causal variants within complex genomic/chromatin landscapes.
幼年特发性关节炎(JIA)患儿外周血细胞的转录组具有明显的转录异常,提示转录调控受损。为了更好地理解这一现象,我们研究了已知的JIA遗传风险位点,其中大多数位于非编码区,在全基因组范围内转录在此受到调控和协调。我们检测了人类中性粒细胞和CD4原代T细胞,以鉴定位于这些风险位点内的基因和功能元件。
我们分析了RNA测序(RNA-Seq)数据、H3K27ac和H3K4me1染色质免疫沉淀测序(ChIP-Seq)数据,以及先前发表的通过双末端标签测序(ChIA-PET)进行的染色质相互作用分析数据,以表征已知的JIA相关风险位点内的染色质景观。
在中性粒细胞和原代CD4+T细胞中,大多数与JIA相关的连锁不平衡(LD)区域都含有H3K27ac和/或H3K4me1标记。这些LD区域也是一小部分转录因子的结合位点,尤其是在中性粒细胞中。此外,这些区域在中性粒细胞中显示出丰富的内含子和基因间转录。在中性粒细胞中,未经治疗的JIA患者与健康儿童之间差异表达的基因均不在JIA风险LD区域内。在CD4+T细胞中,根据ChIA-PET数据确定,多个基因,包括HLA-DQA1、HLA-DQB2、TRAF1和IRF1与LD区域内的长距离相互作用区域相关。
这些发现表明遗传风险导致了JIA中观察到的异常转录控制。此外,这些发现证明了在复杂的基因组/染色质景观中识别实际因果变异的挑战。