Pepke Shirley, Ver Steeg Greg
Lyrid LLC, South Pasadena, USA.
Information Sciences Institute, University of Southern California, Marina Del Rey, USA.
BMC Med Genomics. 2017 Mar 15;10(1):12. doi: 10.1186/s12920-017-0245-6.
De novo inference of clinically relevant gene function relationships from tumor RNA-seq remains a challenging task. Current methods typically either partition patient samples into a few subtypes or rely upon analysis of pairwise gene correlations that will miss some groups in noisy data. Leveraging higher dimensional information can be expected to increase the power to discern targetable pathways, but this is commonly thought to be an intractable computational problem.
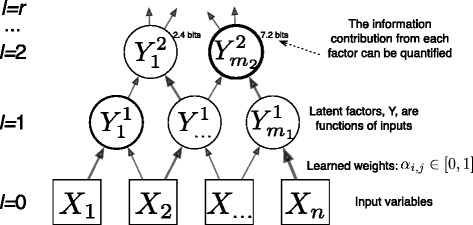
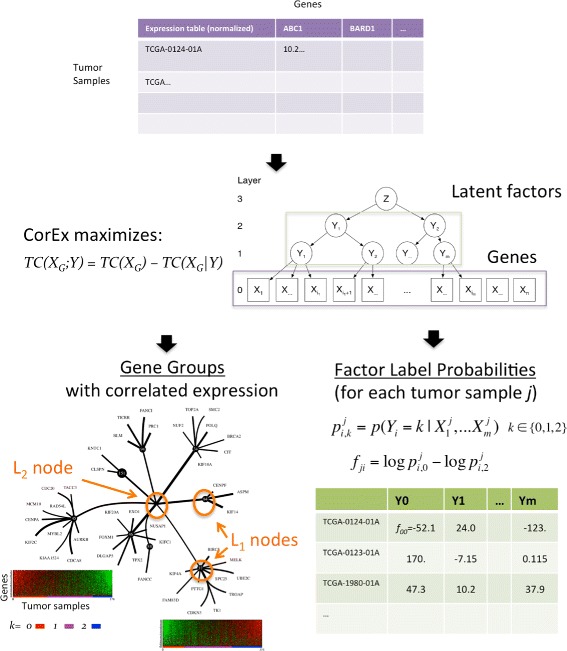
In this work we adapt a recently developed machine learning algorithm for sensitive detection of complex gene relationships. The algorithm, CorEx, efficiently optimizes over multivariate mutual information and can be iteratively applied to generate a hierarchy of relatively independent latent factors. The learned latent factors are used to stratify patients for survival analysis with respect to both single factors and combinations. These analyses are performed and interpreted in the context of biological function annotations and protein network interactions that might be utilized to match patients to multiple therapies.
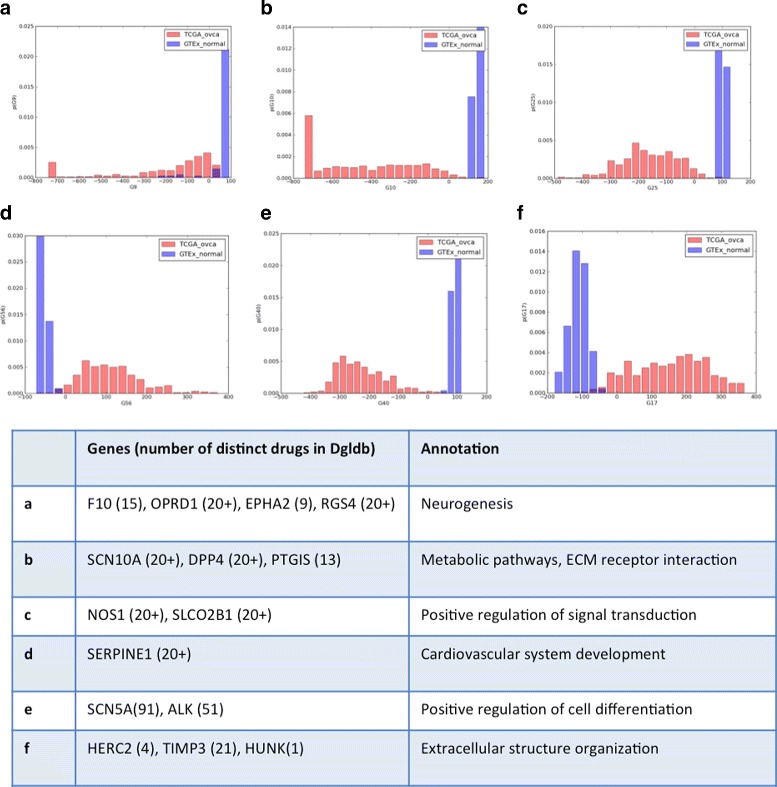
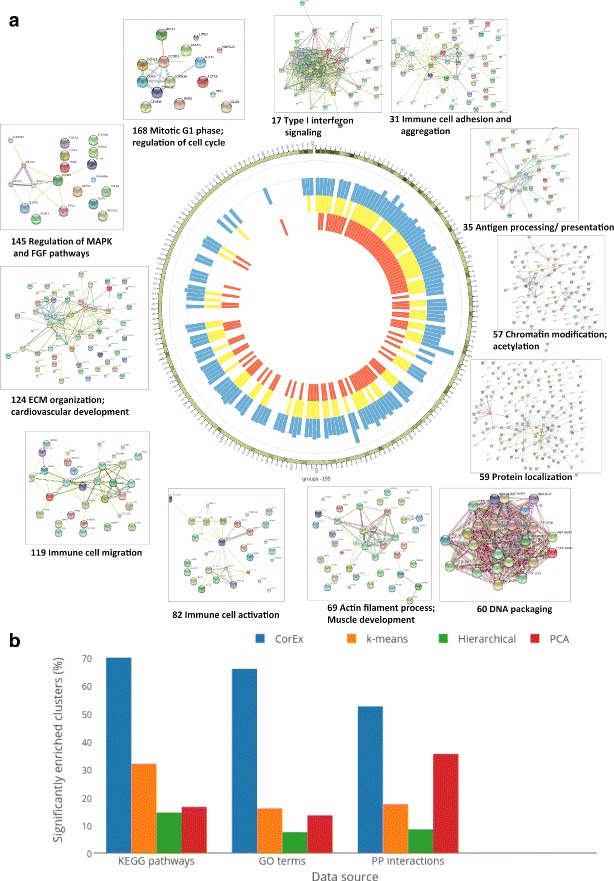
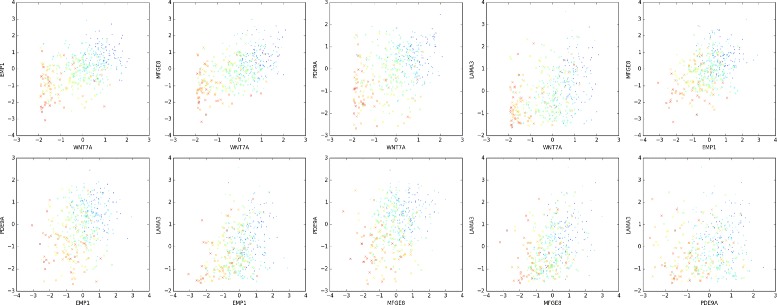
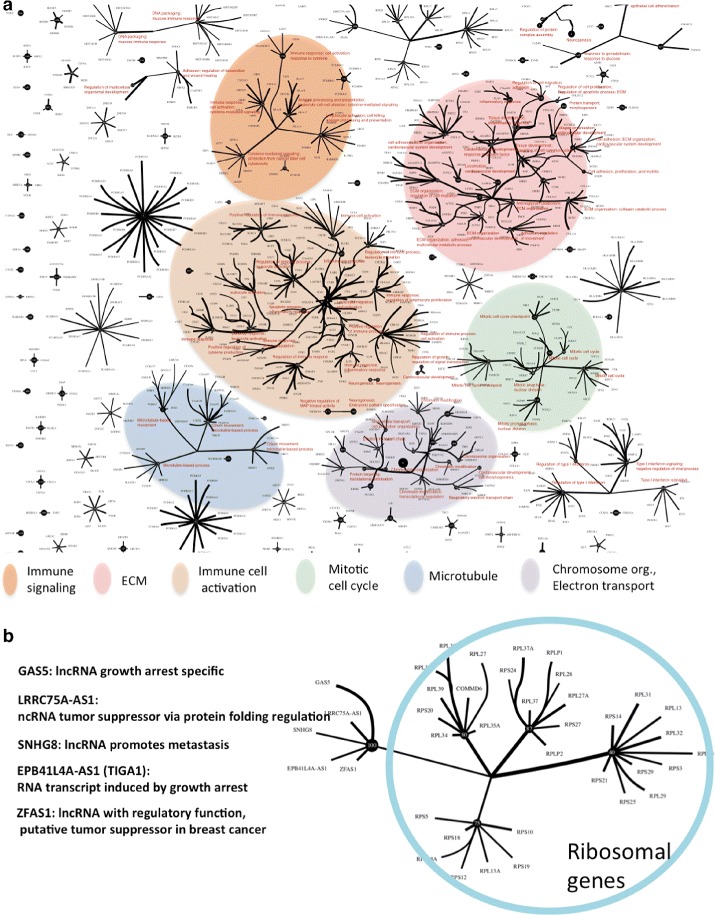
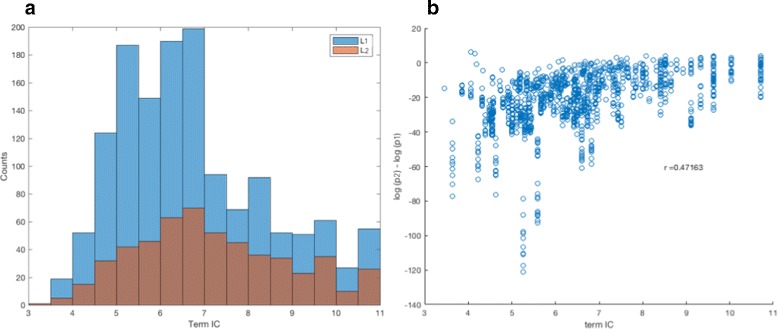
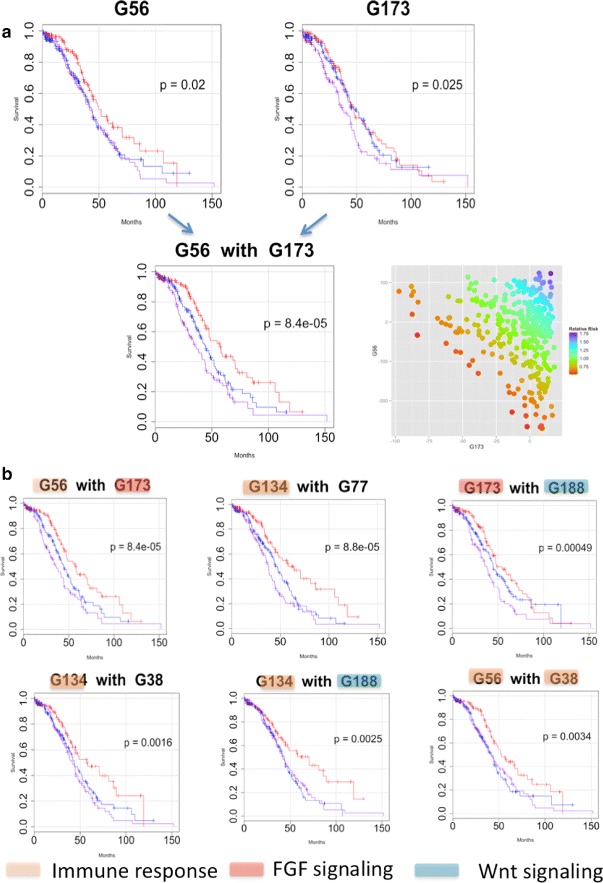
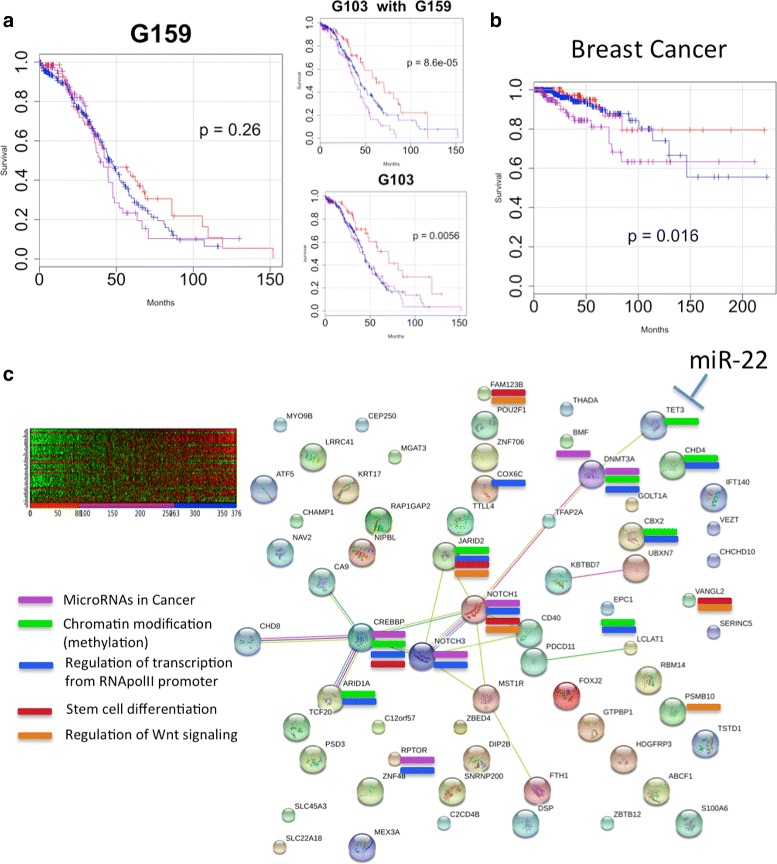
Analysis of ovarian tumor RNA-seq samples demonstrates the algorithm's power to infer well over one hundred biologically interpretable gene cohorts, several times more than standard methods such as hierarchical clustering and k-means. The CorEx factor hierarchy is also informative, with related but distinct gene clusters grouped by upper nodes. Some latent factors correlate with patient survival, including one for a pathway connected with the epithelial-mesenchymal transition in breast cancer that is regulated by a microRNA that modulates epigenetics. Further, combinations of factors lead to a synergistic survival advantage in some cases.
In contrast to studies that attempt to partition patients into a small number of subtypes (typically 4 or fewer) for treatment purposes, our approach utilizes subgroup information for combinatoric transcriptional phenotyping. Considering only the 66 gene expression groups that are found to both have significant Gene Ontology enrichment and are small enough to indicate specific drug targets implies a computational phenotype for ovarian cancer that allows for 3 possible patient profiles, enabling truly personalized treatment. The findings here demonstrate a new technique that sheds light on the complexity of gene expression dependencies in tumors and could eventually enable the use of patient RNA-seq profiles for selection of personalized and effective cancer treatments.
从肿瘤RNA测序中重新推断临床相关基因功能关系仍然是一项具有挑战性的任务。当前方法通常要么将患者样本划分为少数几种亚型,要么依赖于成对基因相关性分析,而这种分析会在噪声数据中遗漏一些组。利用更高维度的信息有望提高识别可靶向通路的能力,但人们普遍认为这是一个难以解决的计算问题。
在这项工作中,我们采用了一种最近开发的机器学习算法来灵敏地检测复杂的基因关系。该算法CorEx在多变量互信息上进行高效优化,并且可以迭代应用以生成相对独立的潜在因子层次结构。所学习到的潜在因子用于对患者进行分层,以便就单一因素及其组合进行生存分析。这些分析是在生物学功能注释和蛋白质网络相互作用的背景下进行的,这些注释和相互作用可用于将患者与多种治疗方法进行匹配。
对卵巢肿瘤RNA测序样本的分析表明该算法能够推断出超过一百个具有生物学可解释性的基因群组,比诸如层次聚类和k均值等标准方法多出几倍。CorEx因子层次结构也具有信息性,相关但不同的基因簇由上层节点分组。一些潜在因子与患者生存相关,包括一个与乳腺癌上皮-间质转化相关的通路,该通路由一种调节表观遗传学的微小RNA调控。此外,在某些情况下,因子组合会带来协同的生存优势。
与试图为治疗目的将患者划分为少数几种亚型(通常为4种或更少)的研究不同,我们的方法利用亚组信息进行组合转录表型分析。仅考虑发现既具有显著基因本体富集且又足够小以指示特定药物靶点的66个基因表达组,就意味着卵巢癌的一种计算表型,该表型允许3种可能的患者概况,从而实现真正的个性化治疗。此处的研究结果展示了一种新技术,该技术揭示了肿瘤中基因表达依赖性的复杂性,并最终可能使患者RNA测序概况用于选择个性化且有效的癌症治疗成为可能。