Wang Yuwei, Han Rui, Zhang Huimin, Liu Hongli, Li Jiazhong, Liu Huanxiang, Gramatica Paola
School of Pharmacy, Lanzhou University, 199 West Donggang Rd., Lanzhou 730000, China.
Department of Theoretical and Applied Sciences, University of Insubria, Via Dunant 3, 21100 Varese, Italy.
Biomed Res Int. 2017;2017:3572394. doi: 10.1155/2017/3572394. Epub 2017 Feb 15.
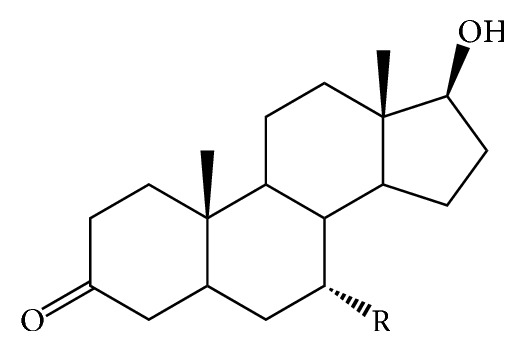
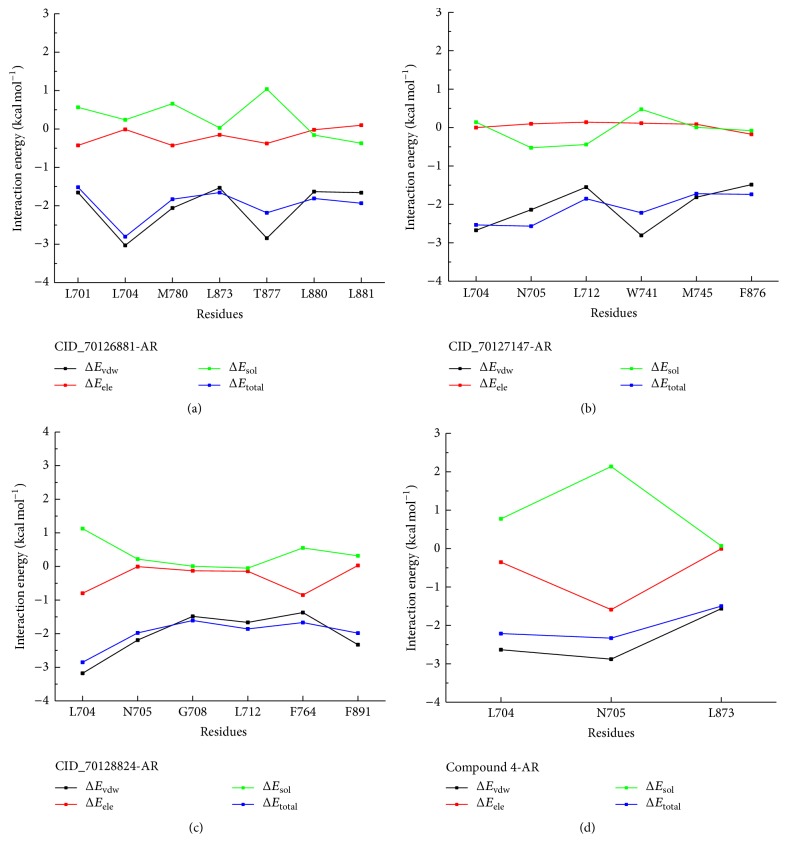
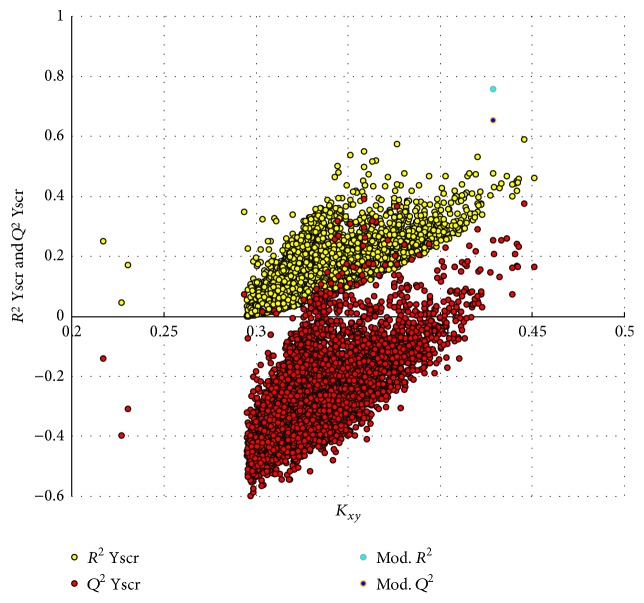
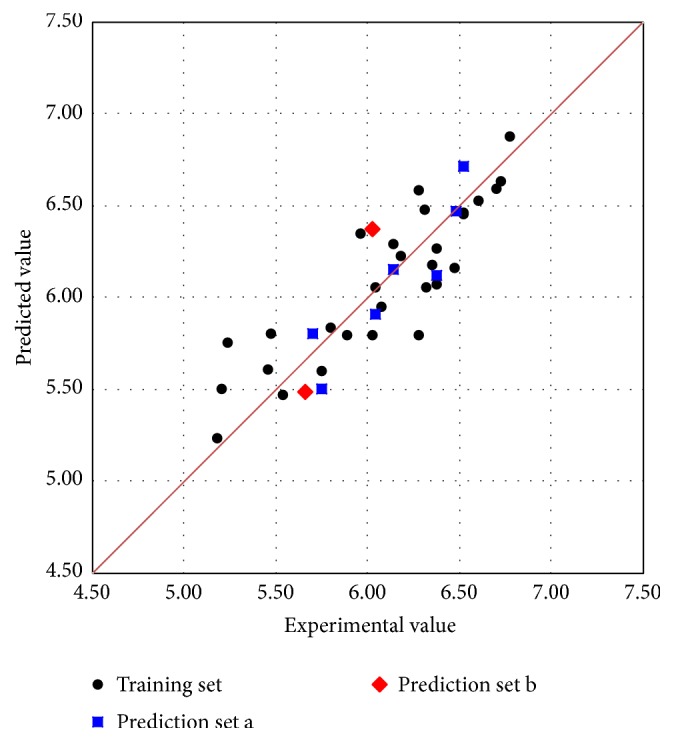
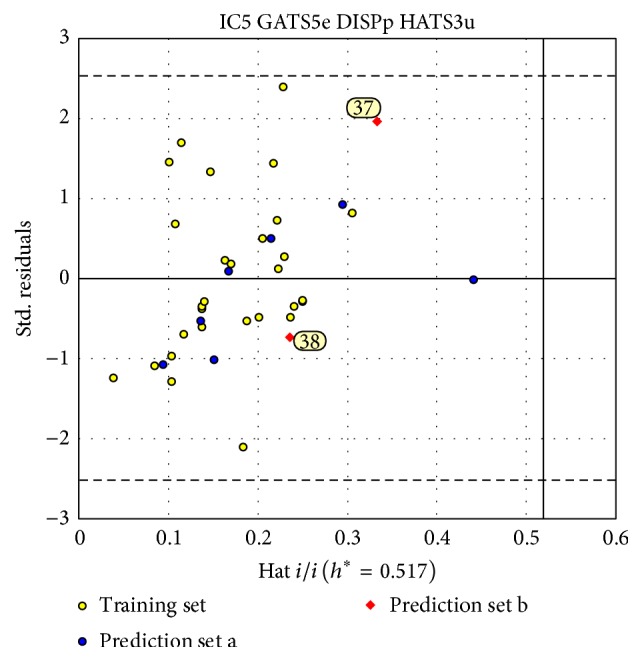
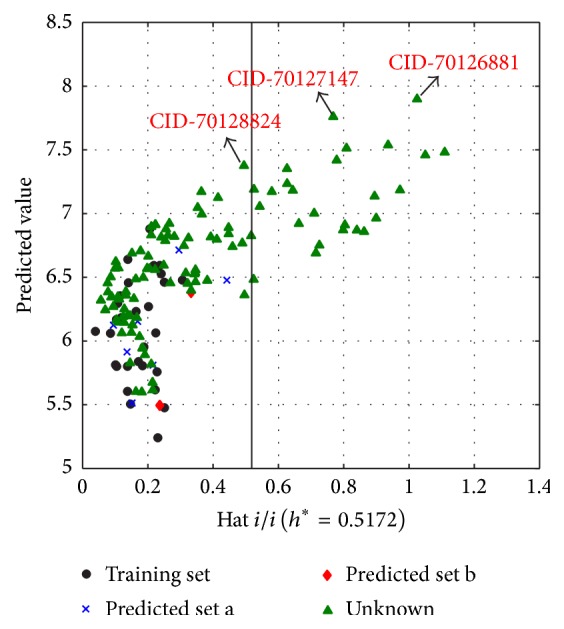
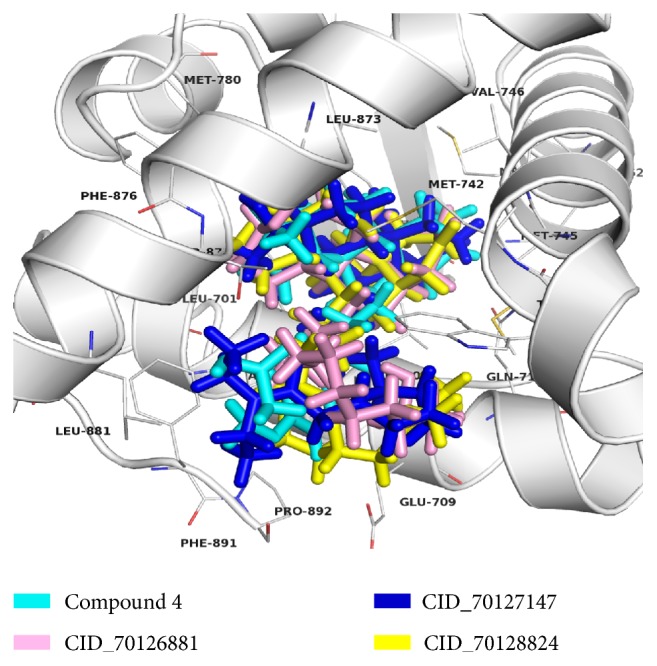
The antiandrogens, such as bicalutamide, targeting the androgen receptor (AR), are the main endocrine therapies for prostate cancer (PCa). But as drug resistance to antiandrogens emerges in advanced PCa, there presents a high medical need for exploitation of novel AR antagonists. In this work, the relationships between the molecular structures and antiandrogenic activities of a series of 7-substituted dihydrotestosterone derivatives were investigated. The proposed MLR model obtained high predictive ability. The thoroughly validated QSAR model was used to virtually screen new dihydrotestosterones derivatives taken from PubChem, resulting in the finding of novel compounds CID_70128824, CID_70127147, and CID_70126881, whose in silico bioactivities are much higher than the published best one, even higher than bicalutamide. In addition, molecular docking, molecular dynamics (MD) simulations, and MM/GBSA have been employed to analyze and compare the binding modes between the novel compounds and AR. Through the analysis of the binding free energy and residue energy decomposition, we concluded that the newly discovered chemicals can in silico bind to AR with similar position and mechanism to the reported active compound and the van der Waals interaction is the main driving force during the binding process.
抗雄激素药物,如比卡鲁胺,通过作用于雄激素受体(AR)发挥作用,是前列腺癌(PCa)的主要内分泌治疗药物。然而,随着晚期PCa对抗雄激素药物产生耐药性,开发新型AR拮抗剂的医疗需求变得极为迫切。在本研究中,我们探究了一系列7-取代二氢睾酮衍生物的分子结构与抗雄激素活性之间的关系。所构建的多元线性回归(MLR)模型展现出了较高的预测能力。经过全面验证的定量构效关系(QSAR)模型被用于从PubChem数据库中虚拟筛选新的二氢睾酮衍生物,从而发现了新型化合物CID_70128824、CID_70127147和CID_70126881,它们的计算机模拟生物活性远高于已发表的最佳化合物,甚至超过了比卡鲁胺。此外,我们还运用分子对接、分子动力学(MD)模拟以及MM/GBSA方法,对新型化合物与AR之间的结合模式进行了分析和比较。通过对结合自由能和残基能量分解的分析,我们得出结论:新发现的化合物在计算机模拟中能够与AR结合,其结合位置和机制与已报道的活性化合物相似,范德华相互作用是结合过程中的主要驱动力。