Li Xiujin, Lund Mogens Sandø, Janss Luc, Wang Chonglong, Ding Xiangdong, Zhang Qin, Su Guosheng
Center for Quantitative Genetics and Genomics, Department of Molecular Biology and Genetics, Aarhus University, Tjele, Denmark.
Laboratory of Animal Genetics, Breeding and Reproduction, Ministry of Agriculture of China, National Engineering Laboratory for Animal Breeding, College of Animal Science and Technology, China Agricultural University, Beijing, 100193, China.
BMC Genet. 2017 Mar 15;18(1):26. doi: 10.1186/s12863-017-0491-9.
With the development of SNP chips, SNP information provides an efficient approach to further disentangle different patterns of genomic variances and covariances across the genome for traits of interest. Due to the interaction between genotype and environment as well as possible differences in genetic background, it is reasonable to treat the performances of a biological trait in different populations as different but genetic correlated traits. In the present study, we performed an investigation on the patterns of region-specific genomic variances, covariances and correlations between Chinese and Nordic Holstein populations for three milk production traits.
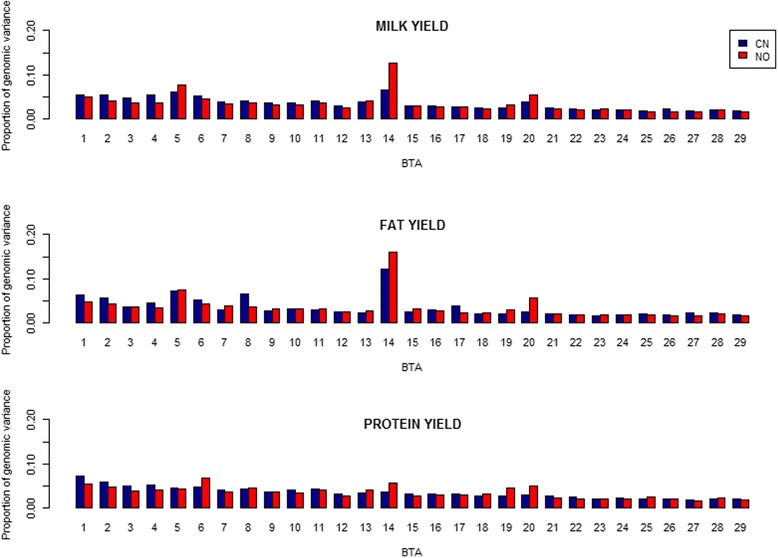
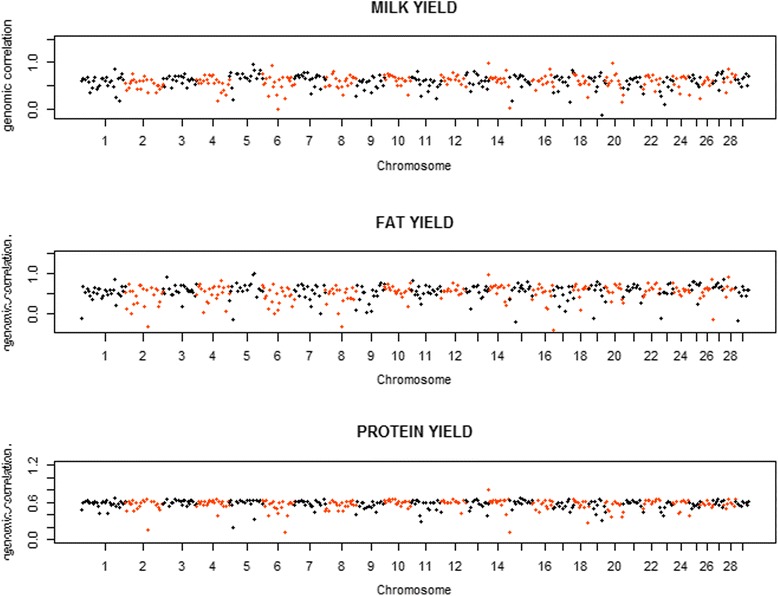
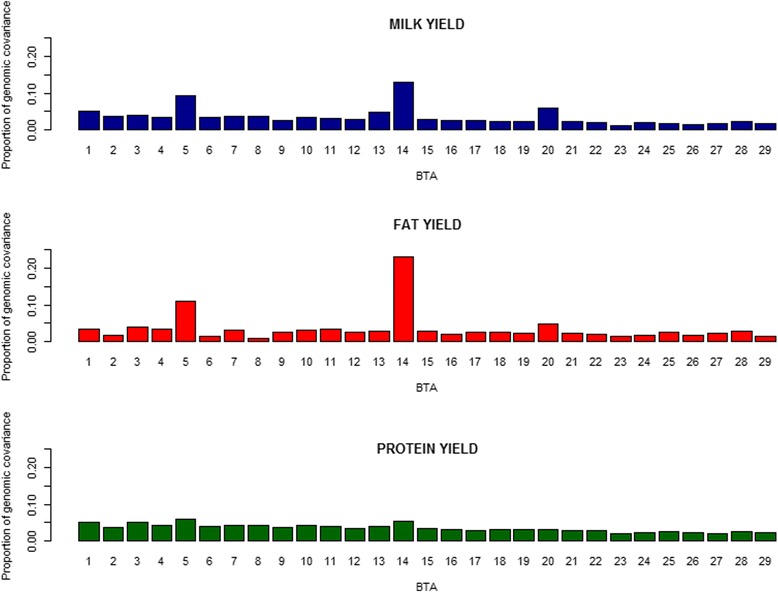
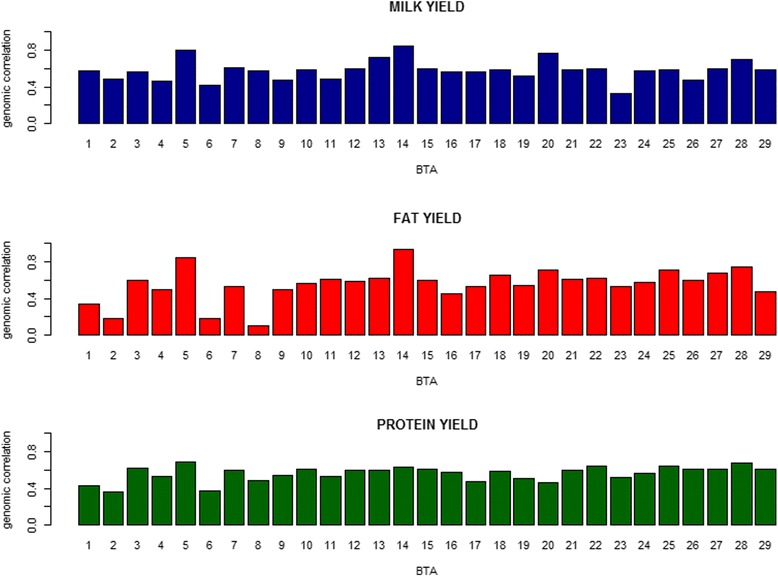
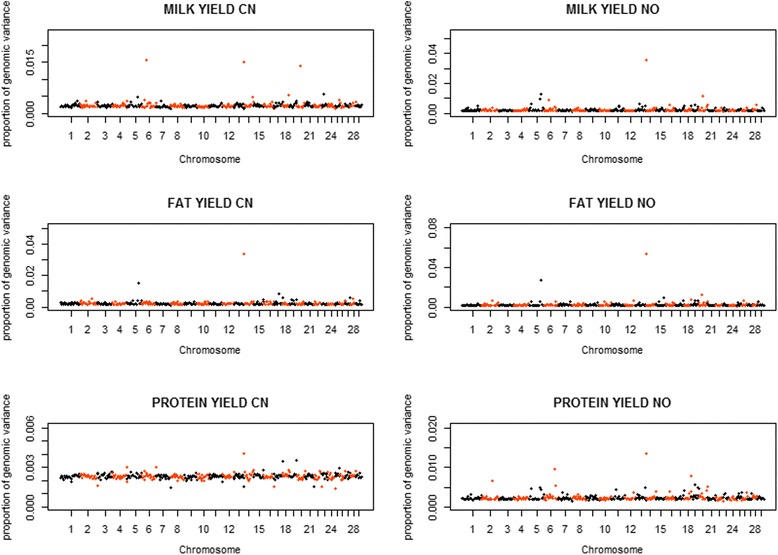
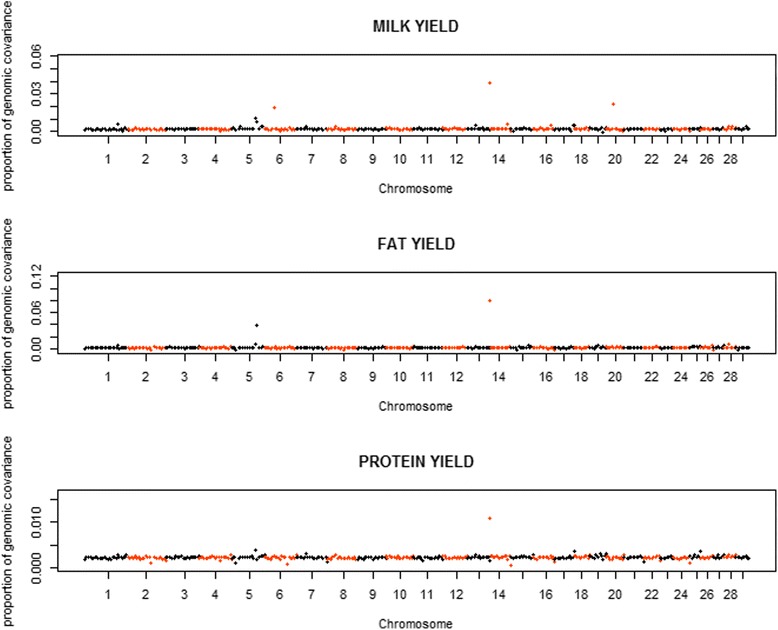
Variances and covariances between Chinese and Nordic Holstein populations were estimated for genomic regions at three different levels of genome region (all SNP as one region, each chromosome as one region and every 100 SNP as one region) using a novel multi-trait random regression model which uses latent variables to model heterogeneous variance and covariance. In the scenario of the whole genome as one region, the genomic variances, covariances and correlations obtained from the new multi-trait Bayesian method were comparable to those obtained from a multi-trait GBLUP for all the three milk production traits. In the scenario of each chromosome as one region, BTA 14 and BTA 5 accounted for very large genomic variance, covariance and correlation for milk yield and fat yield, whereas no specific chromosome showed very large genomic variance, covariance and correlation for protein yield. In the scenario of every 100 SNP as one region, most regions explained <0.50% of genomic variance and covariance for milk yield and fat yield, and explained <0.30% for protein yield, while some regions could present large variance and covariance. Although overall correlations between two populations for the three traits were positive and high, a few regions still showed weakly positive or highly negative genomic correlations for milk yield and fat yield.
The new multi-trait Bayesian method using latent variables to model heterogeneous variance and covariance could work well for estimating the genomic variances and covariances for all genome regions simultaneously. Those estimated genomic parameters could be useful to improve the genomic prediction accuracy for Chinese and Nordic Holstein populations using a joint reference data in the future.
随着单核苷酸多态性(SNP)芯片的发展,SNP信息为进一步解析全基因组中感兴趣性状的不同基因组方差和协方差模式提供了一种有效方法。由于基因型与环境之间的相互作用以及遗传背景可能存在的差异,将生物性状在不同群体中的表现视为不同但具有遗传相关性的性状是合理的。在本研究中,我们针对中国荷斯坦牛和北欧荷斯坦牛群体的三个产奶性状,对区域特异性基因组方差、协方差和相关性模式进行了调查。
使用一种新颖的多性状随机回归模型,该模型利用潜在变量对异质方差和协方差进行建模,在基因组区域的三个不同水平(将所有SNP作为一个区域、将每条染色体作为一个区域以及每100个SNP作为一个区域)估计了中国荷斯坦牛和北欧荷斯坦牛群体之间的方差和协方差。在将整个基因组作为一个区域的情况下,新的多性状贝叶斯方法获得的基因组方差、协方差和相关性与多性状基因组最佳线性无偏预测(GBLUP)方法针对所有三个产奶性状获得的结果相当。在将每条染色体作为一个区域的情况下,对于产奶量和乳脂率,第14号牛染色体(BTA 14)和第5号牛染色体(BTA 5)占非常大的基因组方差、协方差和相关性,而对于乳蛋白率,没有特定染色体表现出非常大的基因组方差、协方差和相关性。在每100个SNP作为一个区域的情况下,大多数区域对产奶量和乳脂率的基因组方差和协方差的解释率<0.50%,对乳蛋白率的解释率<0.30%,而一些区域可能呈现较大的方差和协方差。尽管两个群体在这三个性状上的总体相关性为正且较高,但仍有一些区域对产奶量和乳脂率表现出弱正或高负的基因组相关性。
使用潜在变量对异质方差和协方差进行建模的新多性状贝叶斯方法能够很好地同时估计所有基因组区域的基因组方差和协方差。这些估计的基因组参数可能有助于未来通过联合参考数据提高中国荷斯坦牛和北欧荷斯坦牛群体的基因组预测准确性。