Department of Molecular Biology and Genetics, Center for Quantitative Genetics and Genomics, Aarhus University, Blichers Allé 20, P.O. Box 50, 8830, Tjele, Denmark.
Animal Breeding and Genomics Centre, Wageningen University, PO Box 338, 6700 AH, Wageningen, The Netherlands.
Genet Sel Evol. 2017 Dec 5;49(1):89. doi: 10.1186/s12711-017-0364-8.
Accurate genomic prediction requires a large reference population, which is problematic for traits that are expensive to measure. Traits related to milk protein composition are not routinely recorded due to costly procedures and are considered to be controlled by a few quantitative trait loci of large effect. The amount of variation explained may vary between regions leading to heterogeneous (co)variance patterns across the genome. Genomic prediction models that can efficiently take such heterogeneity of (co)variances into account can result in improved prediction reliability. In this study, we developed and implemented novel univariate and bivariate Bayesian prediction models, based on estimates of heterogeneous (co)variances for genome segments (BayesAS). Available data consisted of milk protein composition traits measured on cows and de-regressed proofs of total protein yield derived for bulls. Single-nucleotide polymorphisms (SNPs), from 50K SNP arrays, were grouped into non-overlapping genome segments. A segment was defined as one SNP, or a group of 50, 100, or 200 adjacent SNPs, or one chromosome, or the whole genome. Traditional univariate and bivariate genomic best linear unbiased prediction (GBLUP) models were also run for comparison. Reliabilities were calculated through a resampling strategy and using deterministic formula.
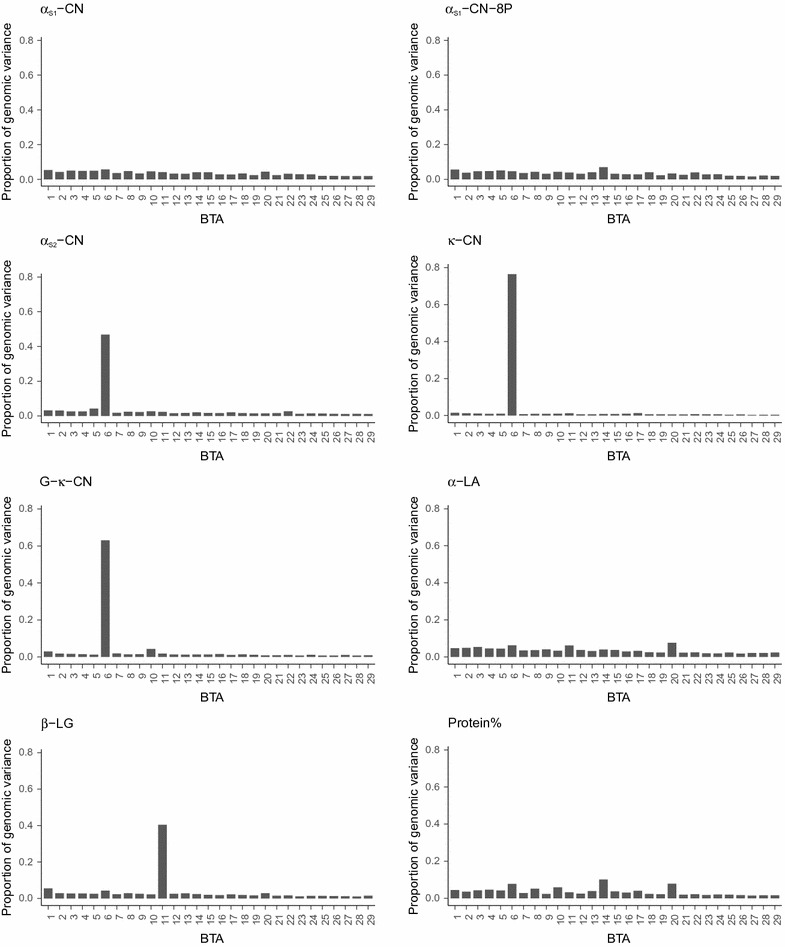
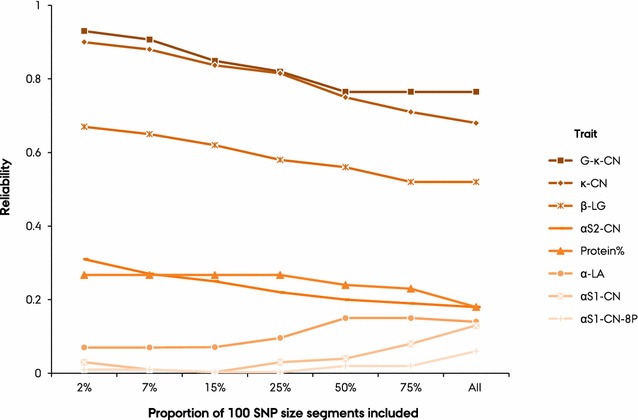
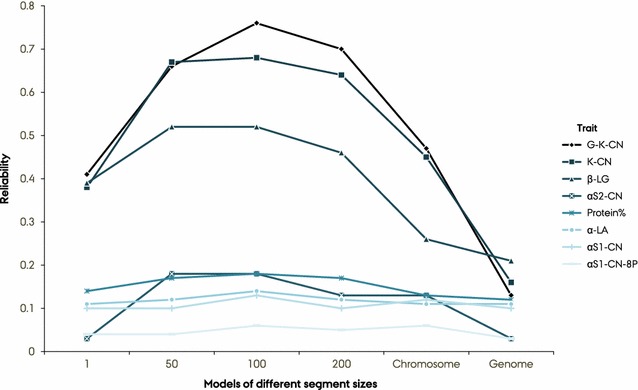
BayesAS models improved prediction reliability for most of the traits compared to GBLUP models and this gain depended on segment size and genetic architecture of the traits. The gain in prediction reliability was especially marked for the protein composition traits β-CN, κ-CN and β-LG, for which prediction reliabilities were improved by 49 percentage points on average using the MT-BayesAS model with a 100-SNP segment size compared to the bivariate GBLUP. Prediction reliabilities were highest with the BayesAS model that uses a 100-SNP segment size. The bivariate versions of our BayesAS models resulted in extra gains of up to 6% in prediction reliability compared to the univariate versions.
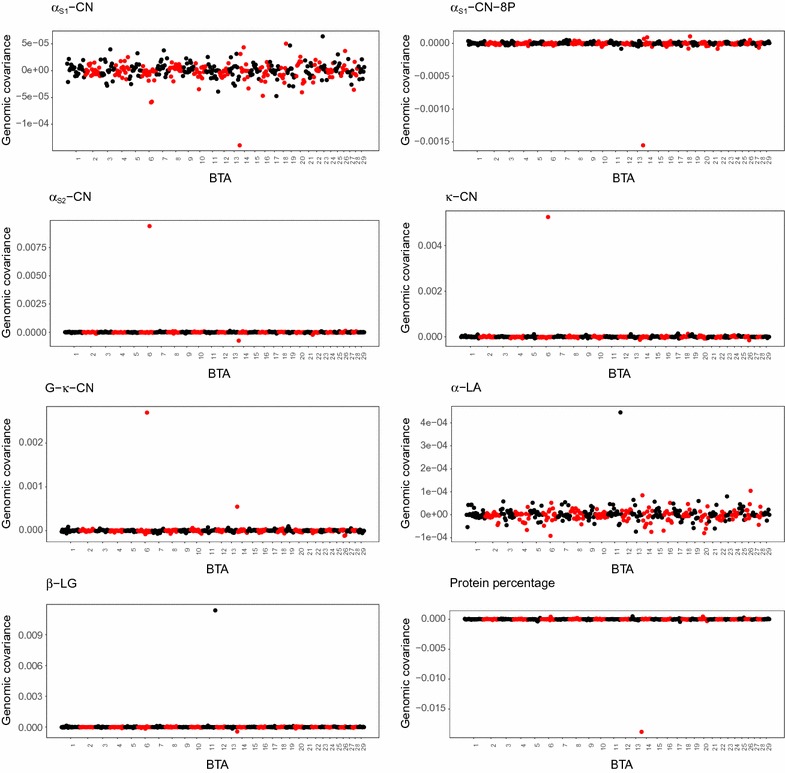
Substantial improvement in prediction reliability was possible for most of the traits related to milk protein composition using our novel BayesAS models. Grouping adjacent SNPs into segments provided enhanced information to estimate parameters and allowing the segments to have different (co)variances helped disentangle heterogeneous (co)variances across the genome.
准确的基因组预测需要一个大型的参考群体,但对于那些测量成本较高的性状来说,这是一个问题。与牛奶蛋白组成相关的性状由于成本较高的程序而没有被常规记录,并且被认为是由少数几个具有较大效应的数量性状位点控制的。在不同区域,解释的变异量可能会有所不同,从而导致基因组中(协)方差的异质性模式。能够有效地考虑到(协)方差这种异质性的基因组预测模型可以提高预测的可靠性。在这项研究中,我们开发并实施了新的单变量和双变量贝叶斯预测模型,这些模型是基于对基因组片段(BayesAS)异质(协)方差的估计。可用数据包括对奶牛进行的牛奶蛋白组成性状的测量值和为公牛推导的去回归总蛋白产量的证明。单核苷酸多态性(SNP)来自 50K SNP 数组,被分组为非重叠的基因组片段。一个片段定义为一个 SNP,或者一组 50、100 或 200 个相邻的 SNP,或者一个染色体,或者整个基因组。还运行了传统的单变量和双变量基因组最佳线性无偏预测(GBLUP)模型进行比较。可靠性是通过重采样策略和使用确定性公式计算的。
与 GBLUP 模型相比,BayesAS 模型提高了大多数性状的预测可靠性,而且这种增益取决于片段大小和性状的遗传结构。对于β-CN、κ-CN 和β-LG 等蛋白质组成性状,增益尤其显著,与使用 100-SNP 片段大小的 MT-BayesAS 模型相比,预测可靠性平均提高了 49 个百分点,而与双变量 GBLUP 相比。使用 100-SNP 片段大小的 BayesAS 模型可获得最高的预测可靠性。与单变量版本相比,我们的 BayesAS 模型的双变量版本在预测可靠性方面额外提高了高达 6%。
使用我们的新 BayesAS 模型,与牛奶蛋白组成相关的大多数性状的预测可靠性都有了显著提高。将相邻的 SNP 分组到片段中可以提供增强的信息来估计参数,并且允许片段具有不同的(协)方差有助于解开基因组中的异质(协)方差。