Nguyen Linh, Dang Cuong C, Ballester Pedro J
Cancer Research Center of Marseille, INSERM U1068, Marseille, France; Institut Paoli-Calmettes, Marseille, France; Aix-Marseille Université, Marseille, France; Cancer Research Center of Marseille UMR7258, Marseille, France.
F1000Res. 2016 Dec 28;5. doi: 10.12688/f1000research.10529.2. eCollection 2016.
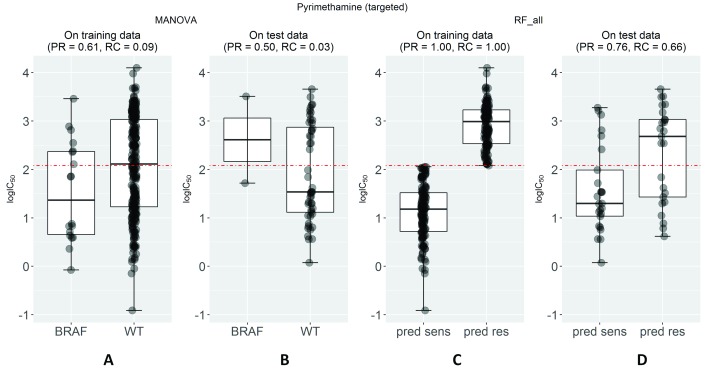
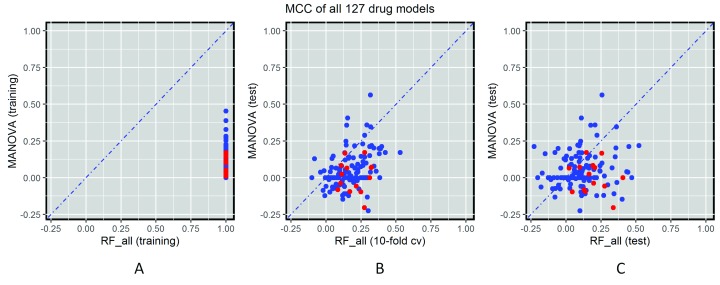
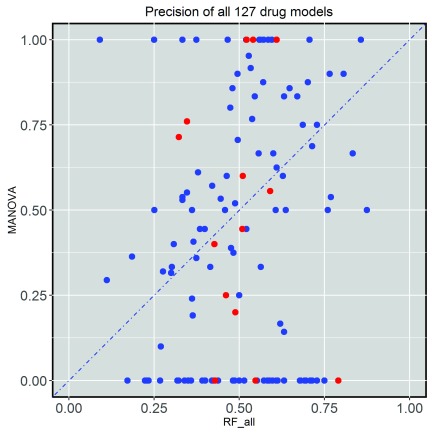
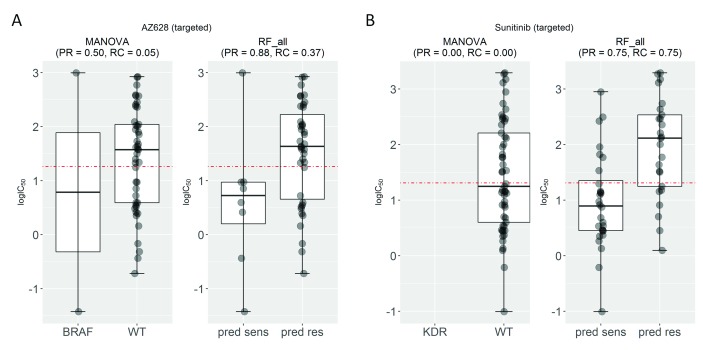
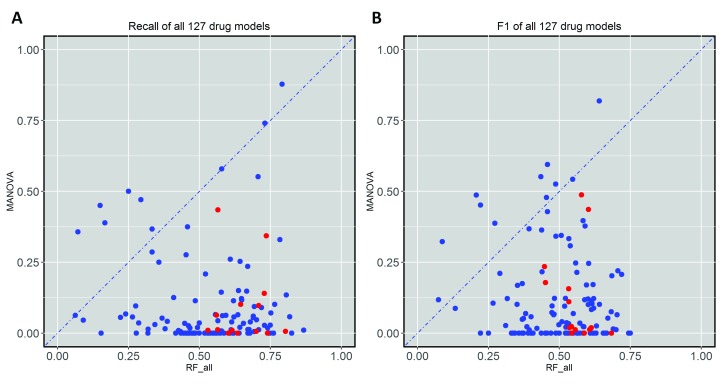
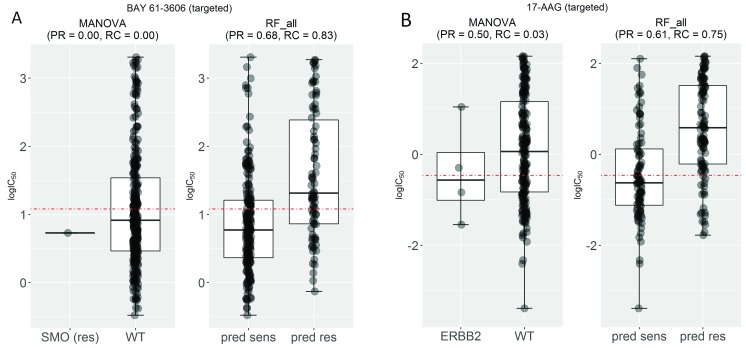
Selected gene mutations are routinely used to guide the selection of cancer drugs for a given patient tumour. Large pharmacogenomic data sets, such as those by Genomics of Drug Sensitivity in Cancer (GDSC) consortium, were introduced to discover more of these single-gene markers of drug sensitivity. Very recently, machine learning regression has been used to investigate how well cancer cell line sensitivity to drugs is predicted depending on the type of molecular profile. The latter has revealed that gene expression data is the most predictive profile in the pan-cancer setting. However, no study to date has exploited GDSC data to systematically compare the performance of machine learning models based on multi-gene expression data against that of widely-used single-gene markers based on genomics data. Here we present this systematic comparison using Random Forest (RF) classifiers exploiting the expression levels of 13,321 genes and an average of 501 tested cell lines per drug. To account for time-dependent batch effects in IC measurements, we employ independent test sets generated with more recent GDSC data than that used to train the predictors and show that this is a more realistic validation than standard k-fold cross-validation. Across 127 GDSC drugs, our results show that the single-gene markers unveiled by the MANOVA analysis tend to achieve higher precision than these RF-based multi-gene models, at the cost of generally having a poor recall (i.e. correctly detecting only a small part of the cell lines sensitive to the drug). Regarding overall classification performance, about two thirds of the drugs are better predicted by the multi-gene RF classifiers. Among the drugs with the most predictive of these models, we found pyrimethamine, sunitinib and 17-AAG. Thanks to this unbiased validation, we now know that this type of models can predict tumour response to some of these drugs. These models can thus be further investigated on tumour models. R code to facilitate the construction of alternative machine learning models and their validation in the presented benchmark is available at http://ballester.marseille.inserm.fr/gdsc.transcriptomicDatav2.tar.gz.
特定基因突变常被用于指导为特定患者肿瘤选择癌症药物。大型药物基因组数据集,如癌症药物敏感性基因组学(GDSC)联盟提供的数据集,被引入以发现更多此类药物敏感性单基因标记。最近,机器学习回归已被用于研究根据分子图谱类型预测癌细胞系对药物敏感性的效果如何。后者表明基因表达数据在泛癌背景下是最具预测性的图谱。然而,迄今为止,尚无研究利用GDSC数据系统地比较基于多基因表达数据的机器学习模型与基于基因组学数据的广泛使用的单基因标记的性能。在此,我们使用随机森林(RF)分类器进行了这种系统比较,该分类器利用13321个基因的表达水平以及每种药物平均501个测试细胞系。为了考虑IC测量中的时间依赖性批次效应,我们使用比用于训练预测器的GDSC数据更新的独立测试集,并表明这是比标准k折交叉验证更现实的验证。在127种GDSC药物中,我们的结果表明,MANOVA分析揭示的单基因标记往往比这些基于RF的多基因模型具有更高的精度,但代价通常是召回率较低(即只能正确检测出对药物敏感的细胞系中的一小部分)。关于整体分类性能,约三分之二的药物由多基因RF分类器预测效果更好。在这些模型预测性最强的药物中,我们发现了乙胺嘧啶、舒尼替尼和17-AAG。由于这种无偏验证,我们现在知道这类模型可以预测肿瘤对其中一些药物的反应。因此,可以在肿瘤模型上进一步研究这些模型。用于促进构建替代机器学习模型及其在本文基准测试中进行验证的R代码可在http://ballester.marseille.inserm.fr/gdsc.transcriptomicDatav2.tar.gz获取。