Seo Ji-Hui, Kang Ilnam, Yang Seung-Jo, Cho Jang-Cheon
Department of Biological Sciences, Inha University, Incheon, Republic of Korea.
PLoS One. 2017 Mar 17;12(3):e0174159. doi: 10.1371/journal.pone.0174159. eCollection 2017.
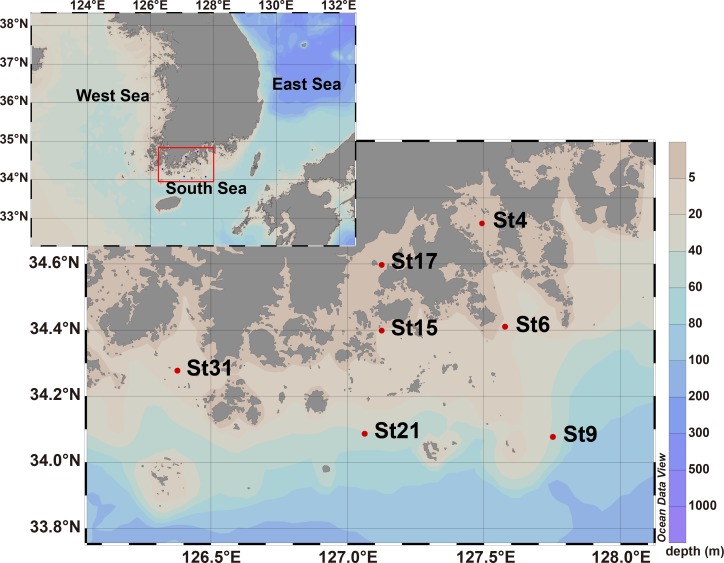
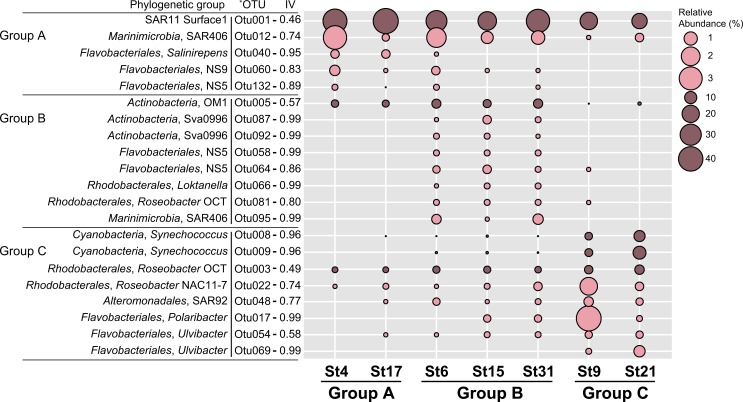
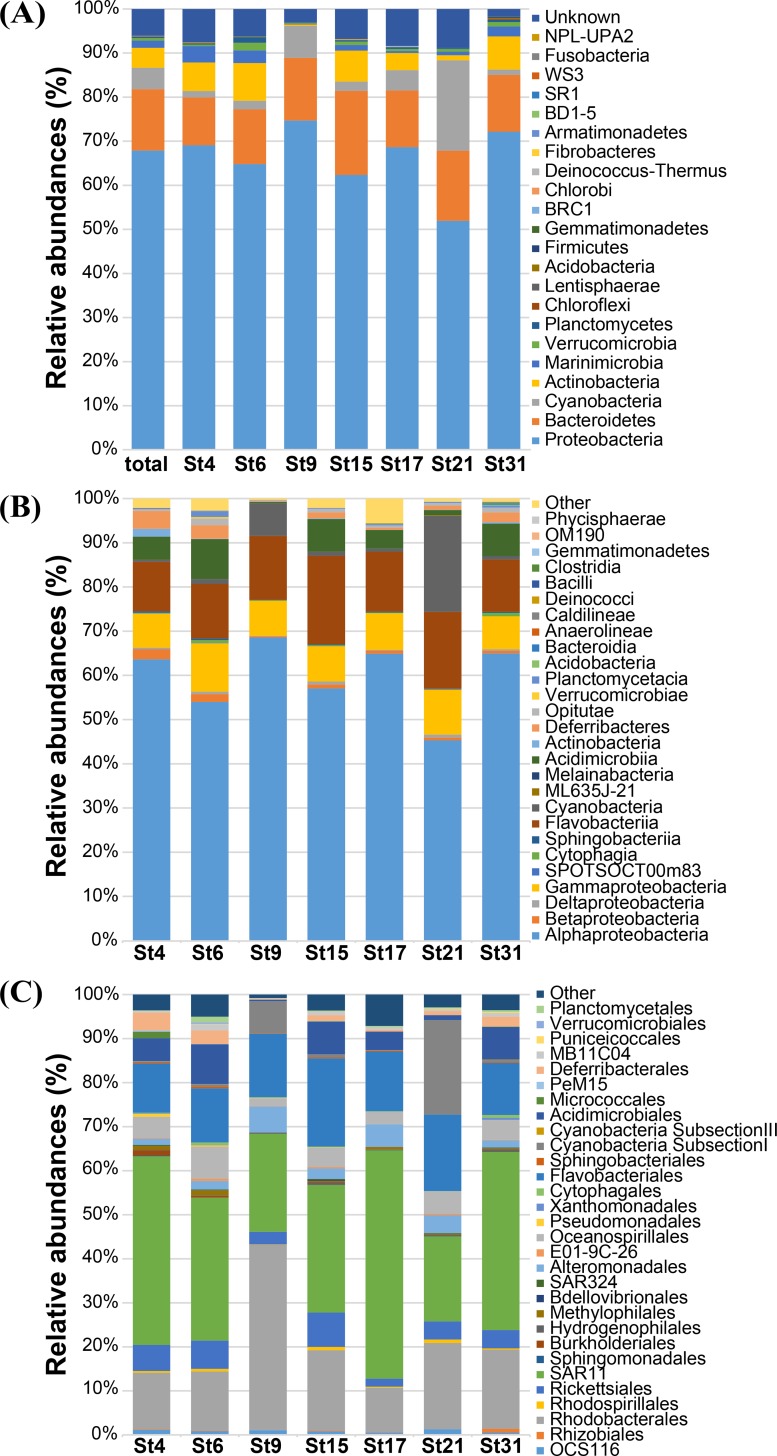
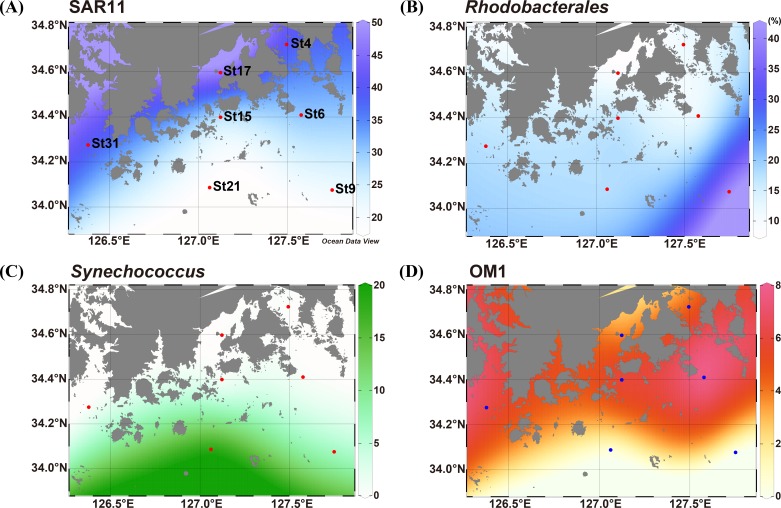
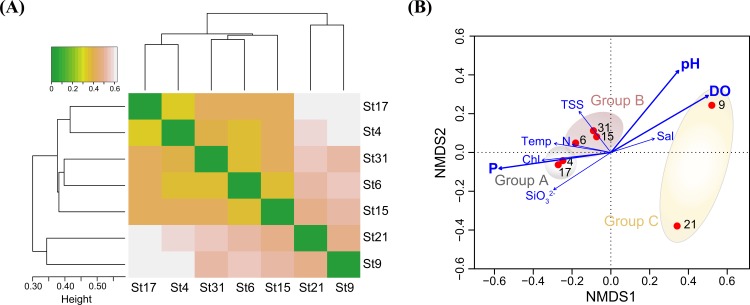
In order to investigate the importance of spatial and environmental factors on the structure and diversity of bacterial communities, high-resolution 16S rRNA gene tag pyrosequencing was applied to bacterial communities in the littoral sea. Seawater samples were prepared from seven different stations in the South Sea of Korea, the marginal sea in the western Pacific Ocean, and were divided into three groups according to distances from the coastline. The majority of 19,860 sequences were affiliated with Alphaproteobacteria (58.2%), Gammaproteobacteria (7.9%), and Bacteroidetes (13.9%). The bacterioplankton community at each station was highly diverse and varied among the samples. Major bacterial lineages showed different niche preferences among three locational groups. Alphaproteobacteria was the most abundant bacterial class, and it harbored the most frequently recorded operational taxonomic units (OTUs) in all sampling stations. However, dominant groups at the order levels showed a clear difference among the samples. The SAR11 clade was more abundant in coastal waters while the Roseobacter clade prevailed at stations far away from the coastline. Furthermore, members of Actinobacteria and Cyanobacteria also exhibited spatial variability. The OM1 clade in Actinobacteria constituted a predominant fraction in coastal samples, but it was essentially absent at the distal stations closer to open ocean. In contrast, Synechococcus was the predominant taxon in the distal samples, accounting for 7.1-19.5%, but was hardly detected in coastal waters, representing less than 0.7%. In Bacteroidetes, NS5 and NS9 groups tended to inhabit coastal waters while the genera Polaribacter and Ulvibacter were more abundant in distal stations. Clustering analysis and principle coordinates analysis based on OTU data indicated that bacterial communities in the studied area were separated into three groups that coincided with locational grouping. Statistical analysis showed that phosphate and dissolved oxygen concentration had a significant influence on the bacterial community composition.
为了研究空间和环境因素对细菌群落结构和多样性的重要性,将高分辨率16S rRNA基因标签焦磷酸测序技术应用于滨海海域的细菌群落。海水样本取自韩国南海(西太平洋边缘海)的七个不同站点,并根据离海岸线的距离分为三组。19860条序列中的大部分隶属于变形菌门α-变形菌纲(58.2%)、γ-变形菌纲(7.9%)和拟杆菌门(13.9%)。每个站点的浮游细菌群落高度多样,且样本间存在差异。主要细菌谱系在三个位置组中表现出不同的生态位偏好。α-变形菌纲是最丰富的细菌纲,在所有采样站点中拥有记录最频繁的可操作分类单元(OTU)。然而,样本在目水平上的优势类群存在明显差异。SAR11分支在沿海水域更为丰富,而玫瑰杆菌分支在远离海岸线的站点占主导地位。此外,放线菌门和蓝细菌门的成员也表现出空间变异性。放线菌门中的OM1分支在沿海样本中占主要部分,但在靠近公海的远端站点基本不存在。相反,聚球藻属是远端样本中的主要分类单元,占7.1 - 19.5%,但在沿海水域中几乎检测不到,占比不到0.7%。在拟杆菌门中,NS5和NS9组倾向于栖息在沿海水域,而极地杆菌属和绿弯菌属在远端站点更为丰富。基于OTU数据的聚类分析和主坐标分析表明,研究区域内的细菌群落分为三组,与位置分组一致。统计分析表明,磷酸盐和溶解氧浓度对细菌群落组成有显著影响。