Centre for Bioinformatics, McGill University, Montreal, Quebec, H3G 0B1, Canada.
The Rosalind and Morris Goodman Cancer Research Centre, McGill University, Montreal, Quebec, H3A 1A3, Canada.
Breast Cancer Res. 2017 Mar 21;19(1):32. doi: 10.1186/s13058-017-0824-7.
The ability to reliably identify the state (activated, repressed, or latent) of any molecular process in the tumor of a patient from an individual whole-genome gene expression profile obtained from microarray or RNA sequencing (RNA-seq) promises important clinical utility. Unfortunately, all previous bioinformatics tools are only applicable in large and diverse panels of patients, or are limited to a single specific pathway/process (e.g. proliferation).
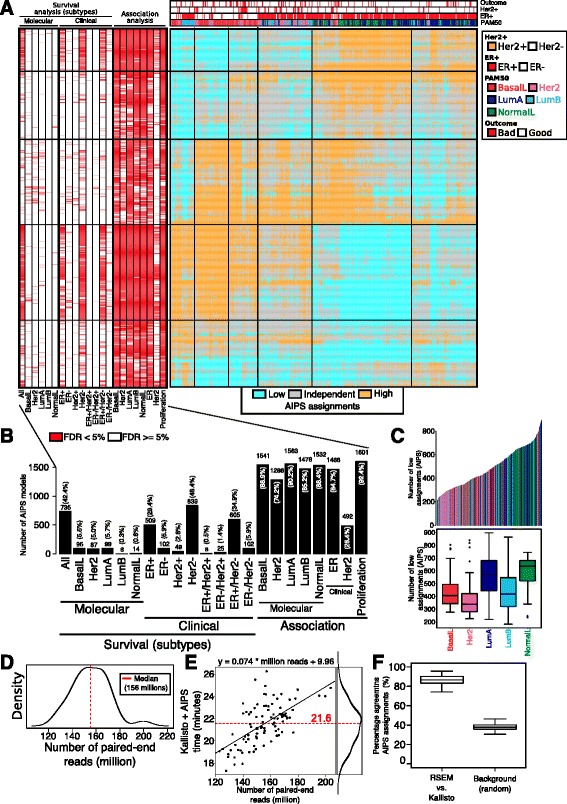
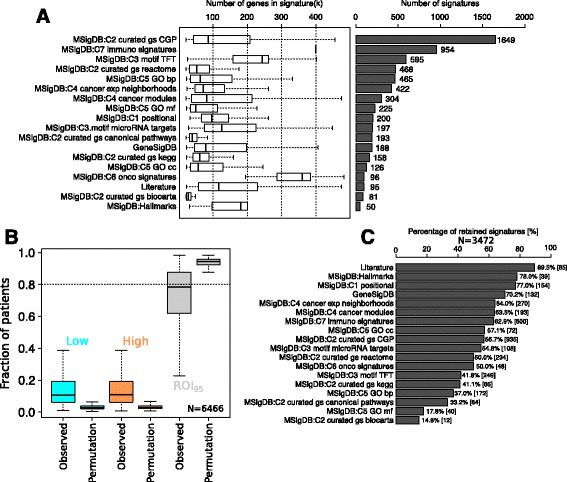
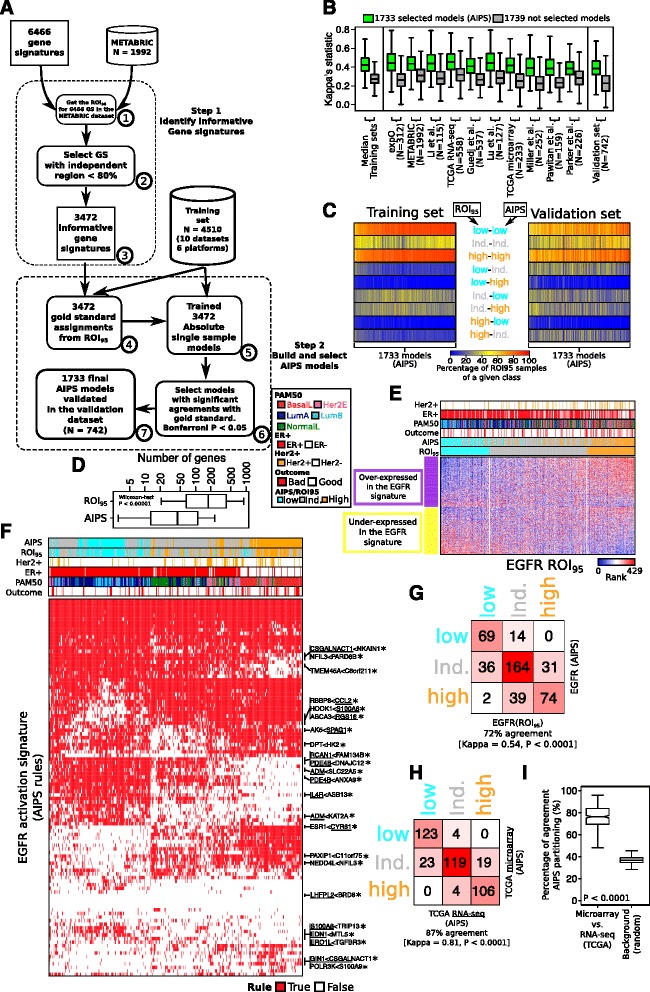
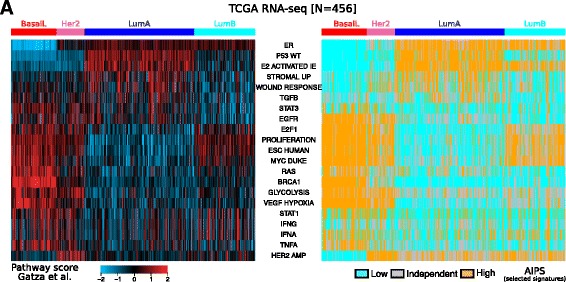
Using a panel of 4510 whole-genome gene expression profiles from 10 different studies we built and selected models predicting the activation status of a compendium of 1733 different biological processes. Using a second independent validation dataset of 742 patients we validated the final list of 1773 models to be included in a de novo tool entitled absolute inference of patient signatures (AIPS). We also evaluated the prognostic significance of the 1773 individual models to predict outcome in all and in specific breast cancer subtypes.
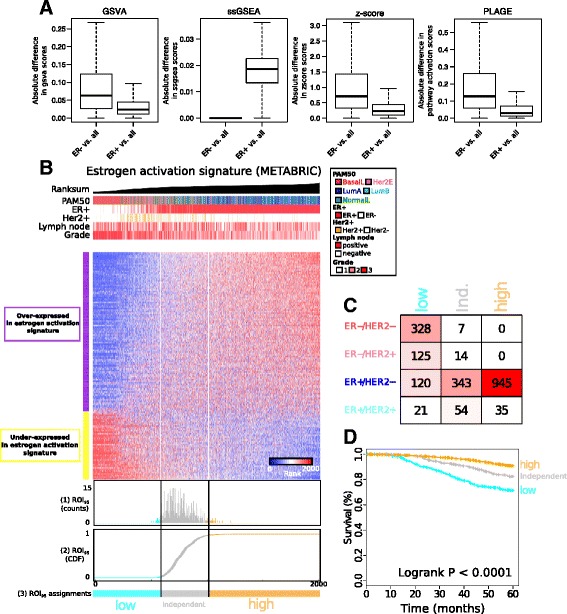
We described the development of the de novo tool entitled AIPS that can identify the activation status of a panel of 1733 different biological processes from an individual breast cancer microarray or RNA-seq profile without recourse to a broad cohort of patients. We demonstrated that AIPS is stable compared to previous tools, as the inferred pathway state is not affected by the composition of a dataset. We also showed that pathway states inferred by AIPS are in agreement with previous tools but use far fewer genes. We determined that several AIPS-defined pathways are prognostic across and within molecularly and clinically define subtypes (two-sided log-rank test false discovery rate (FDR) <5%). Interestingly, 74.5% (1291/1733) of the models are able to distinguish patients with luminal A cancer from those with luminal B cancer (Fisher's exact test FDR <5%).
AIPS represents the first tool that would allow an individual breast cancer patient to obtain a thorough knowledge of the molecular processes active in their tumor from only one individual gene expression (N-of-1) profile.
从微阵列或 RNA 测序 (RNA-seq) 获得的个体全基因组基因表达谱中,可靠地识别患者肿瘤中任何分子过程的状态(激活、抑制或潜伏),有望具有重要的临床应用价值。不幸的是,以前所有的生物信息学工具都仅适用于大量和多样化的患者群体,或者仅限于单一特定途径/过程(例如增殖)。
我们使用来自 10 项不同研究的 4510 个全基因组基因表达谱的面板构建和选择了预测 1733 种不同生物过程激活状态的模型。使用 742 名患者的第二个独立验证数据集,我们验证了最终的 1773 个模型列表,以包含一个名为绝对推断患者特征 (AIPS) 的新工具中。我们还评估了 1773 个个体模型对预测所有和特定乳腺癌亚型结局的预后意义。
我们描述了一种名为 AIPS 的新工具的开发,该工具可以从单个乳腺癌微阵列或 RNA-seq 谱中识别出 1733 种不同生物过程的激活状态,而无需依赖广泛的患者队列。我们证明 AIPS 比以前的工具更稳定,因为推断的途径状态不受数据集组成的影响。我们还表明,AIPS 推断的途径状态与以前的工具一致,但使用的基因要少得多。我们确定,AIPS 定义的几个途径在分子和临床定义的亚组中具有预后意义(双侧对数秩检验错误发现率 (FDR) <5%)。有趣的是,74.5%(1291/1733)的模型能够区分 luminal A 型癌症和 luminal B 型癌症患者(Fisher 精确检验 FDR <5%)。
AIPS 是第一个工具,它可以使单个乳腺癌患者仅从一个个体基因表达 (N-of-1) 谱中全面了解其肿瘤中活跃的分子过程。