Katz Lee S, Griswold Taylor, Williams-Newkirk Amanda J, Wagner Darlene, Petkau Aaron, Sieffert Cameron, Van Domselaar Gary, Deng Xiangyu, Carleton Heather A
Enteric Diseases Laboratory Branch, Centers for Disease Control and PreventionAtlanta, GA, USA; Center for Food Safety, College of Agricultural and Environmental Sciences, University of GeorgiaGriffin, GA, USA.
Enteric Diseases Laboratory Branch, Centers for Disease Control and PreventionAtlanta, GA, USA; Oak Ridge Institute for Science and Education, Oak Ridge Associated UniversitiesOak Ridge, TN, USA.
Front Microbiol. 2017 Mar 13;8:375. doi: 10.3389/fmicb.2017.00375. eCollection 2017.
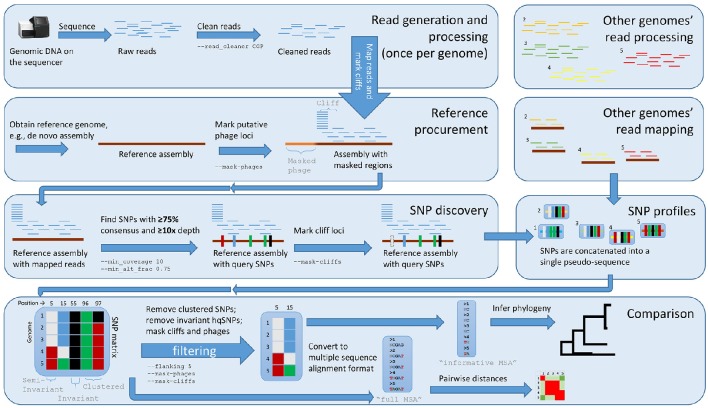
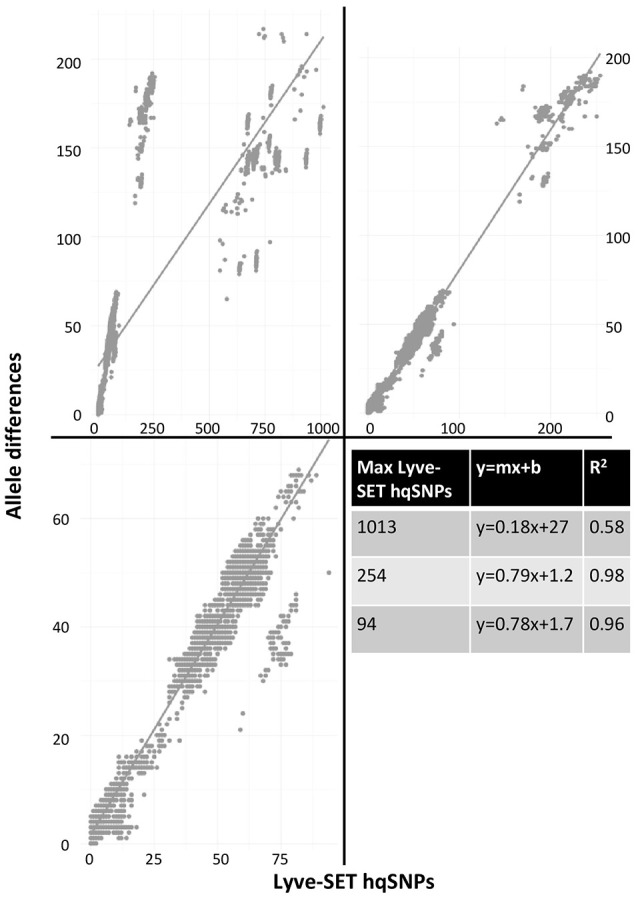
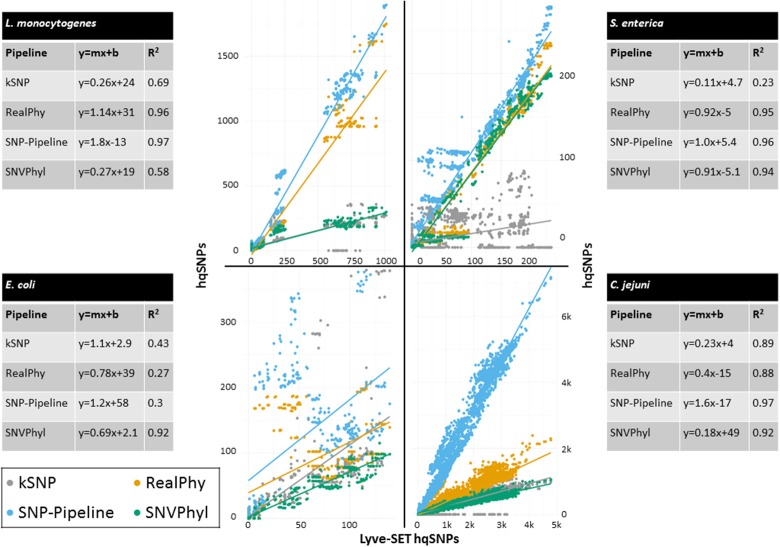
Modern epidemiology of foodborne bacterial pathogens in industrialized countries relies increasingly on whole genome sequencing (WGS) techniques. As opposed to profiling techniques such as pulsed-field gel electrophoresis, WGS requires a variety of computational methods. Since 2013, United States agencies responsible for food safety including the CDC, FDA, and USDA, have been performing whole-genome sequencing (WGS) on all found in clinical, food, and environmental samples. Each year, more genomes of other foodborne pathogens such as , and are being sequenced. Comparing thousands of genomes across an entire species requires a fast method with coarse resolution; however, capturing the fine details of highly related isolates requires a computationally heavy and sophisticated algorithm. Most investigations employing WGS depend on being able to identify an outbreak clade whose inter-genomic distances are less than an empirically determined threshold. When the difference between a few single nucleotide polymorphisms (SNPs) can help distinguish between genomes that are likely outbreak-associated and those that are less likely to be associated, we require a fine-resolution method. To achieve this level of resolution, we have developed Lyve-SET, a high-quality SNP pipeline. We evaluated Lyve-SET by retrospectively investigating 12 outbreak data sets along with four other SNP pipelines that have been used in outbreak investigation or similar scenarios. To compare these pipelines, several distance and phylogeny-based comparison methods were applied, which collectively showed that multiple pipelines were able to identify most outbreak clusters and strains. Currently in the US PulseNet system, whole genome multi-locus sequence typing (wgMLST) is the preferred primary method for foodborne WGS cluster detection and outbreak investigation due to its ability to name standardized genomic profiles, its central database, and its ability to be run in a graphical user interface. However, creating a functional wgMLST scheme requires extended up-front development and subject-matter expertise. When a scheme does not exist or when the highest resolution is needed, SNP analysis is used. Using three outbreak data sets, we demonstrated the concordance between Lyve-SET SNP typing and wgMLST. : Lyve-SET can be found at https://github.com/lskatz/Lyve-SET.
在工业化国家,食源性病原体的现代流行病学越来越依赖全基因组测序(WGS)技术。与脉冲场凝胶电泳等分析技术不同,WGS需要多种计算方法。自2013年以来,美国负责食品安全的机构,包括疾病控制与预防中心(CDC)、食品药品监督管理局(FDA)和美国农业部(USDA),一直在对临床、食品和环境样本中发现的所有[病原体名称未给出]进行全基因组测序。每年,更多其他食源性病原体的基因组,如[病原体名称未给出]、[病原体名称未给出]和[病原体名称未给出],也在被测序。比较整个物种的数千个基因组需要一种具有粗略分辨率的快速方法;然而,捕捉高度相关分离株的精细细节需要计算量大且复杂的算法。大多数采用WGS的[调查名称未给出]依赖于能够识别一个爆发分支,其基因组间距离小于根据经验确定的阈值。当几个单核苷酸多态性(SNP)之间的差异有助于区分可能与爆发相关的基因组和不太可能相关的基因组时,我们需要一种高分辨率方法。为了达到这种分辨率水平,我们开发了Lyve-SET,一种高质量的SNP流程。我们通过回顾性调查12个爆发数据集以及其他四个已用于爆发调查或类似场景的SNP流程来评估Lyve-SET。为了比较这些流程,应用了几种基于距离和系统发育的比较方法,这些方法共同表明多个流程能够识别大多数爆发簇和菌株。目前在美国PulseNet系统中,全基因组多位点序列分型(wgMLST)是食源性WGS簇检测和爆发调查的首选主要方法,因为它能够命名标准化的基因组图谱、其中心数据库以及能够在图形用户界面中运行。然而,创建一个功能性的wgMLST方案需要大量前期开发和专业知识。当不存在方案或需要最高分辨率时,使用SNP分析。使用三个[爆发名称未给出]数据集,我们证明了Lyve-SET SNP分型与wgMLST之间的一致性。:Lyve-SET可在https://github.com/lskatz/Lyve-SET上找到。