Basit Farhan, van Oppen Lisanne Mpe, Schöckel Laura, Bossenbroek Hasse M, van Emst-de Vries Sjenet E, Hermeling Johannes Cw, Grefte Sander, Kopitz Charlotte, Heroult Melanie, Hgm Willems Peter, Koopman Werner Jh
Department of Biochemistry, Radboud Institute for Molecular Life Sciences (RIMLS), Radboud University Medical Centre (Radboudumc), Nijmegen, The Netherlands.
Global Therapeutic Research Group Oncology II, Bayer Pharma AG, Berlin, Germany.
Cell Death Dis. 2017 Mar 30;8(3):e2716. doi: 10.1038/cddis.2017.133.
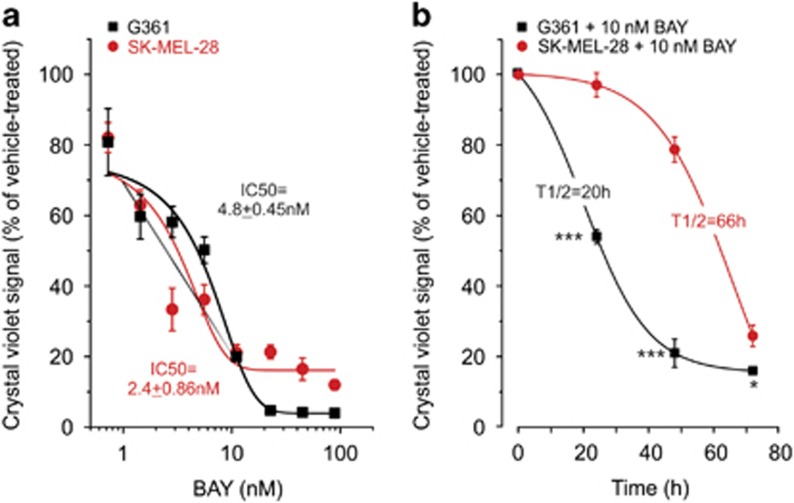
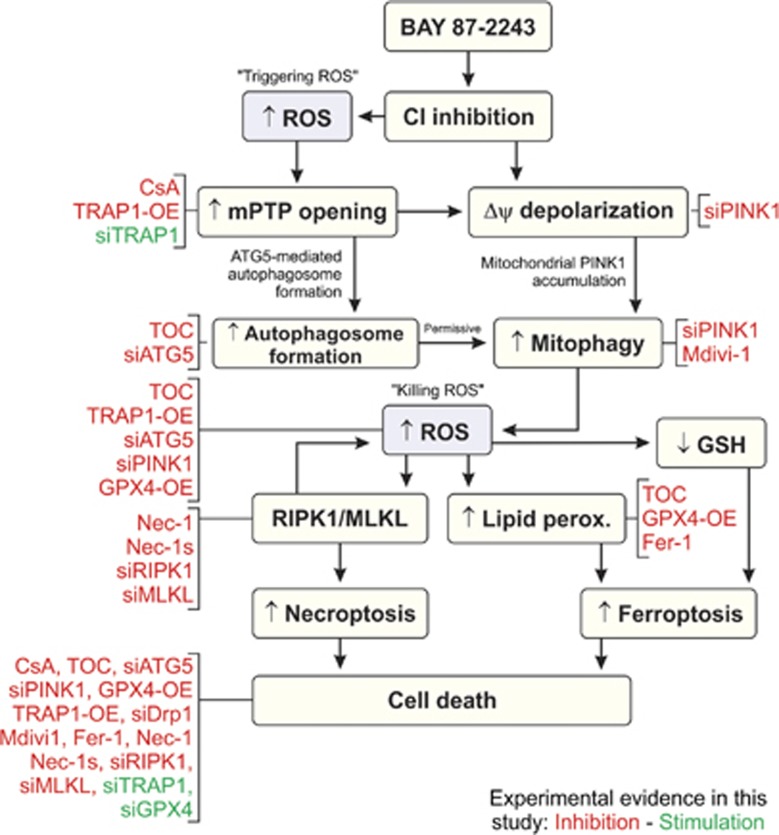
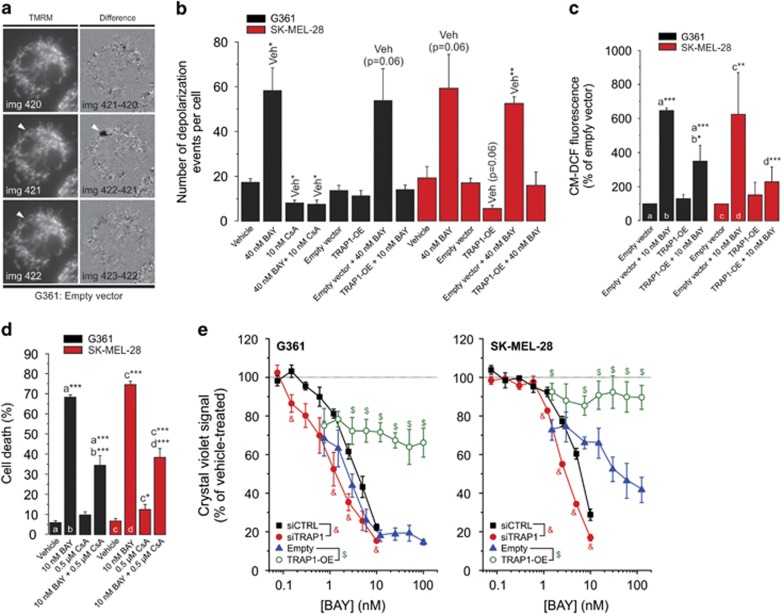
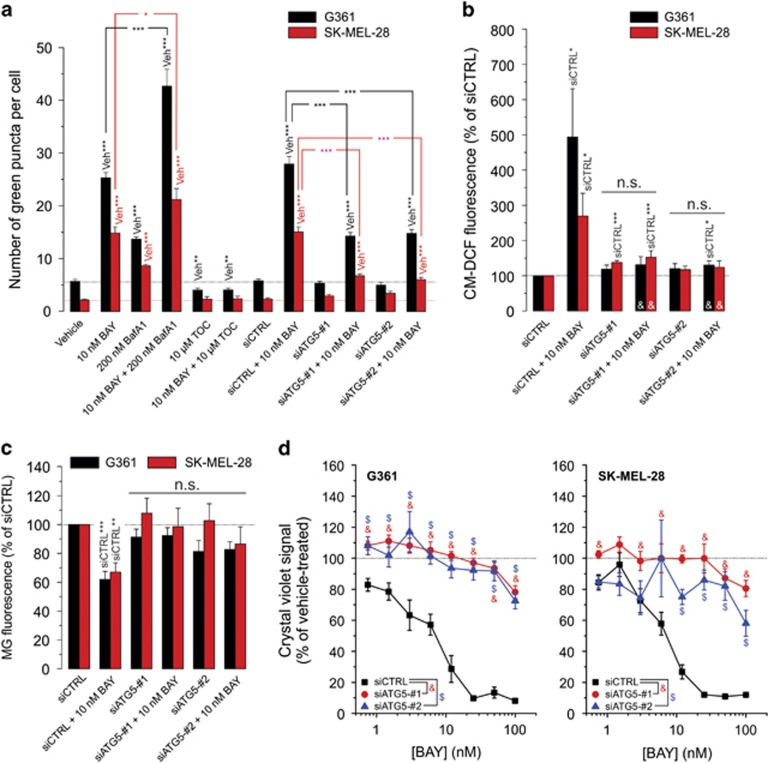
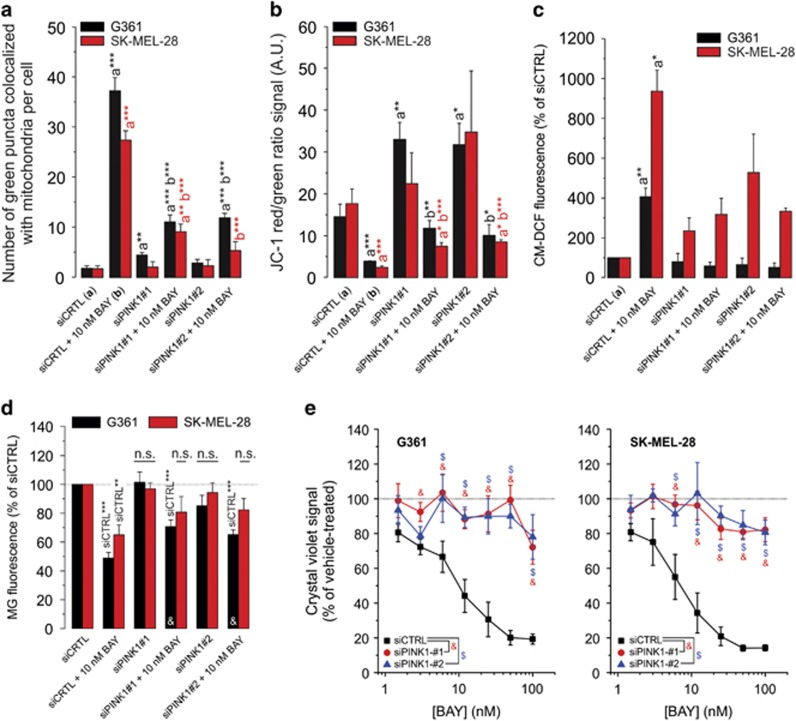
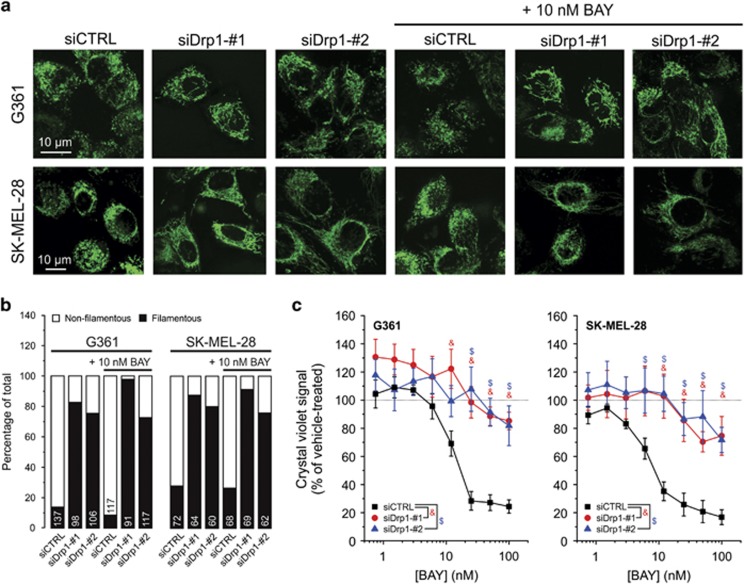
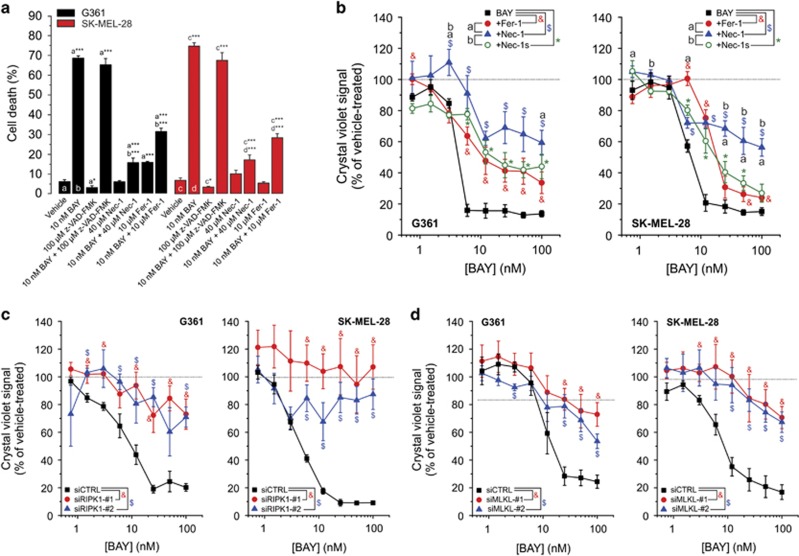
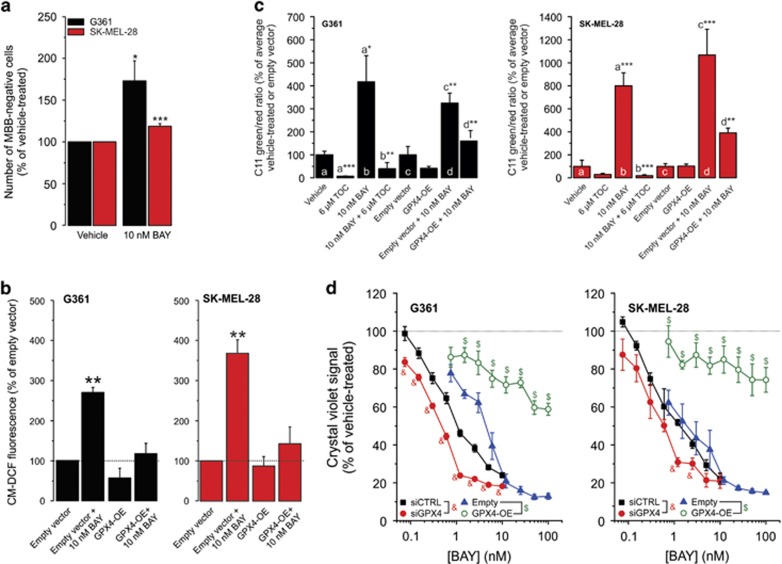
Inhibition of complex I (CI) of the mitochondrial respiratory chain by BAY 87-2243 ('BAY') triggers death of BRAF melanoma cell lines and inhibits in vivo tumor growth. Here we studied the mechanism by which this inhibition induces melanoma cell death. BAY treatment depolarized the mitochondrial membrane potential (Δψ), increased cellular ROS levels, stimulated lipid peroxidation and reduced glutathione levels. These effects were paralleled by increased opening of the mitochondrial permeability transition pore (mPTP) and stimulation of autophagosome formation and mitophagy. BAY-induced cell death was not due to glucose shortage and inhibited by the antioxidant α-tocopherol and the mPTP inhibitor cyclosporin A. Tumor necrosis factor receptor-associated protein 1 (TRAP1) overexpression in BAY-treated cells lowered ROS levels and inhibited mPTP opening and cell death, whereas the latter was potentiated by TRAP1 knockdown. Knockdown of autophagy-related 5 (ATG5) inhibited the BAY-stimulated autophagosome formation, cellular ROS increase and cell death. Knockdown of phosphatase and tensin homolog-induced putative kinase 1 (PINK1) inhibited the BAY-induced Δψ depolarization, mitophagy stimulation, ROS increase and cell death. Dynamin-related protein 1 (Drp1) knockdown induced mitochondrial filamentation and inhibited BAY-induced cell death. The latter was insensitive to the pancaspase inhibitor z-VAD-FMK, but reduced by necroptosis inhibitors (necrostatin-1, necrostatin-1s)) and knockdown of key necroptosis proteins (receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and mixed lineage kinase domain-like (MLKL)). BAY-induced cell death was also reduced by the ferroptosis inhibitor ferrostatin-1 and overexpression of the ferroptosis-inhibiting protein glutathione peroxidase 4 (GPX4). This overexpression also inhibited the BAY-induced ROS increase and lipid peroxidation. Conversely, GPX4 knockdown potentiated BAY-induced cell death. We propose a chain of events in which: (i) CI inhibition induces mPTP opening and Δψ depolarization, that (ii) stimulate autophagosome formation, mitophagy and an associated ROS increase, leading to (iii) activation of combined necroptotic/ferroptotic cell death.
BAY 87-2243(“BAY”)对线粒体呼吸链复合体 I(CI)的抑制作用可触发 BRAF 黑色素瘤细胞系的死亡,并抑制体内肿瘤生长。在此,我们研究了这种抑制作用诱导黑色素瘤细胞死亡的机制。BAY 处理使线粒体膜电位(Δψ)去极化,增加细胞内活性氧(ROS)水平,刺激脂质过氧化并降低谷胱甘肽水平。这些效应伴随着线粒体通透性转换孔(mPTP)开放增加以及自噬体形成和线粒体自噬的刺激。BAY 诱导的细胞死亡并非由于葡萄糖缺乏,且可被抗氧化剂α-生育酚和 mPTP 抑制剂环孢素 A 抑制。在经 BAY 处理的细胞中过表达肿瘤坏死因子受体相关蛋白 1(TRAP1)可降低 ROS 水平,抑制 mPTP 开放和细胞死亡,而敲低 TRAP1 则会增强细胞死亡。敲低自噬相关蛋白 5(ATG5)可抑制 BAY 刺激的自噬体形成、细胞内 ROS 增加和细胞死亡。敲低磷酸酶和张力蛋白同源物诱导的假定激酶 1(PINK1)可抑制 BAY 诱导的Δψ去极化、线粒体自噬刺激、ROS 增加和细胞死亡。动力相关蛋白 1(Drp1)敲低可诱导线粒体成丝,并抑制 BAY 诱导的细胞死亡。后者对泛半胱天冬酶抑制剂 z-VAD-FMK 不敏感,但可被坏死性凋亡抑制剂(坏死性凋亡抑制剂-1、坏死性凋亡抑制剂-1s))以及敲低关键坏死性凋亡蛋白(受体相互作用丝氨酸/苏氨酸蛋白激酶 1(RIPK1)和混合谱系激酶结构域样蛋白(MLKL))所降低。铁死亡抑制剂铁抑素-1 和铁死亡抑制蛋白谷胱甘肽过氧化物酶 4(GPX4)的过表达也可降低 BAY 诱导的细胞死亡。这种过表达还可抑制 BAY 诱导的 ROS 增加和脂质过氧化。相反,敲低 GPX4 会增强 BAY 诱导的细胞死亡。我们提出了一系列事件:(i)CI 抑制诱导 mPTP 开放和Δψ去极化,(ii)刺激自噬体形成、线粒体自噬以及相关的 ROS 增加,导致(iii)联合坏死性凋亡/铁死亡性细胞死亡的激活。