Bansal Vikas
Department of Pediatrics, School of Medicine, University of California San Diego, 9500 Gilman Drive, 92093, La JollaCA, USA.
BMC Bioinformatics. 2017 Mar 14;18(Suppl 3):43. doi: 10.1186/s12859-017-1471-9.
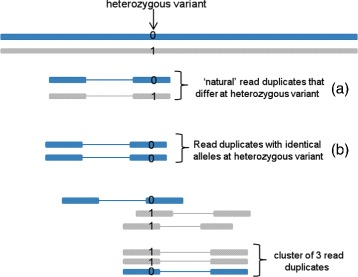
PCR amplification is an important step in the preparation of DNA sequencing libraries prior to high-throughput sequencing. PCR amplification introduces redundant reads in the sequence data and estimating the PCR duplication rate is important to assess the frequency of such reads. Existing computational methods do not distinguish PCR duplicates from "natural" read duplicates that represent independent DNA fragments and therefore, over-estimate the PCR duplication rate for DNA-seq and RNA-seq experiments.
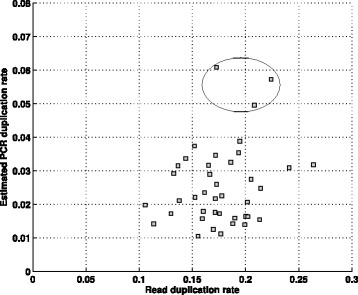
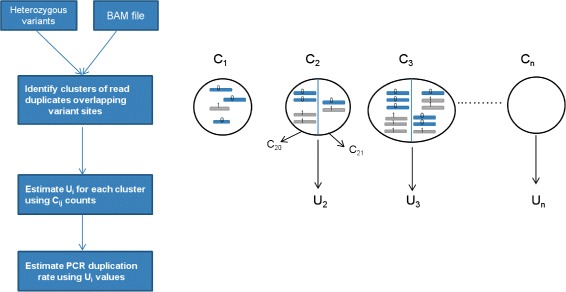
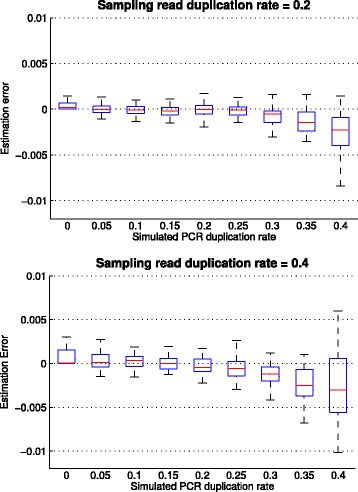
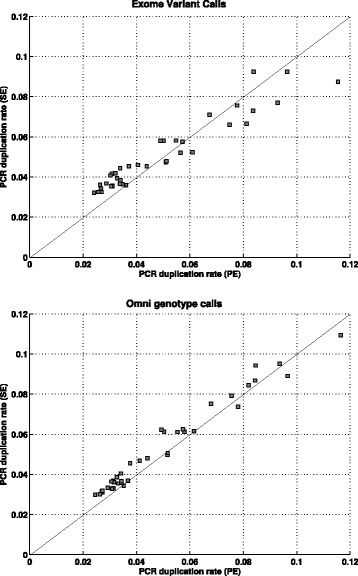
In this paper, we present a computational method to estimate the average PCR duplication rate of high-throughput sequence datasets that accounts for natural read duplicates by leveraging heterozygous variants in an individual genome. Analysis of simulated data and exome sequence data from the 1000 Genomes project demonstrated that our method can accurately estimate the PCR duplication rate on paired-end as well as single-end read datasets which contain a high proportion of natural read duplicates. Further, analysis of exome datasets prepared using the Nextera library preparation method indicated that 45-50% of read duplicates correspond to natural read duplicates likely due to fragmentation bias. Finally, analysis of RNA-seq datasets from individuals in the 1000 Genomes project demonstrated that 70-95% of read duplicates observed in such datasets correspond to natural duplicates sampled from genes with high expression and identified outlier samples with a 2-fold greater PCR duplication rate than other samples.
The method described here is a useful tool for estimating the PCR duplication rate of high-throughput sequence datasets and for assessing the fraction of read duplicates that correspond to natural read duplicates. An implementation of the method is available at https://github.com/vibansal/PCRduplicates .
PCR扩增是高通量测序前DNA测序文库制备中的重要步骤。PCR扩增会在序列数据中引入冗余读段,估计PCR重复率对于评估此类读段的频率很重要。现有的计算方法无法区分PCR重复与代表独立DNA片段的“天然”读段重复,因此会高估DNA测序和RNA测序实验的PCR重复率。
在本文中,我们提出了一种计算方法,通过利用个体基因组中的杂合变异来估计高通量序列数据集的平均PCR重复率,该方法考虑了天然读段重复。对模拟数据和千人基因组计划的外显子组序列数据的分析表明,我们的方法能够准确估计双端及单端读段数据集中的PCR重复率,这些数据集包含高比例的天然读段重复。此外,对使用Nextera文库制备方法制备的外显子组数据集的分析表明,45%-50%的读段重复对应于可能由于片段化偏差导致的天然读段重复。最后,对千人基因组计划中个体的RNA测序数据集的分析表明,在此类数据集中观察到的70%-95%的读段重复对应于从高表达基因中采样的天然重复,并识别出PCR重复率比其他样本高两倍的异常样本。
本文所述方法是估计高通量序列数据集PCR重复率以及评估对应于天然读段重复的读段重复比例的有用工具。该方法的实现可在https://github.com/vibansal/PCRduplicates获取。