Feng Xiao-Dong, Li Li-Wei, Zhang Jian-Hong, Zhu Yun-Ping, Chang Cheng, Shu Kun-Xian, Ma Jie
Chongqing University of Posts and Telecommunications, 2 Chong Wen Road of Nan'an District, Chongqing, 400065, China.
Department of Bioinformatics, State Key Laboratory of Proteomics, Beijing Proteome Research Center, National Engineering Research Center for Protein Drugs, National Center for Protein Sciences (Beijing), Beijing Institute of Radiation Medicine, 38 Life Science Park Road, Beijing, 102206, China.
BMC Genomics. 2017 Mar 14;18(Suppl 2):143. doi: 10.1186/s12864-017-3491-2.
The mass spectrometry based technical pipeline has provided a high-throughput, high-sensitivity and high-resolution platform for post-genomic biology. Varied models and algorithms are implemented by different tools to improve proteomics data analysis. The target-decoy searching strategy has become the most popular strategy to control false identification in peptide and protein identifications. While this strategy can estimate the false discovery rate (FDR) within a dataset, it cannot directly evaluate the false positive matches in target identifications.
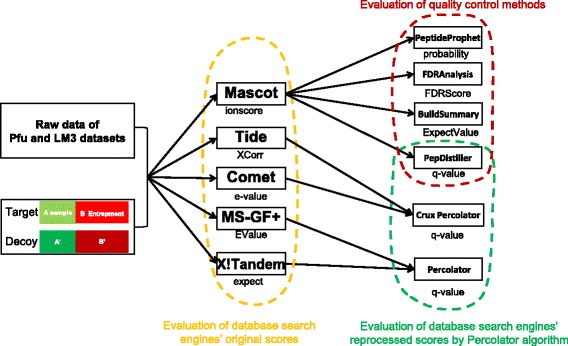
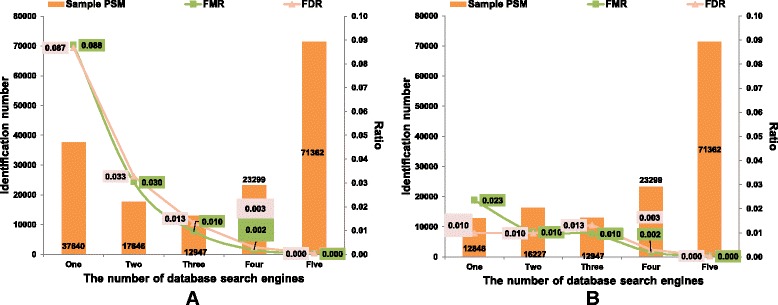
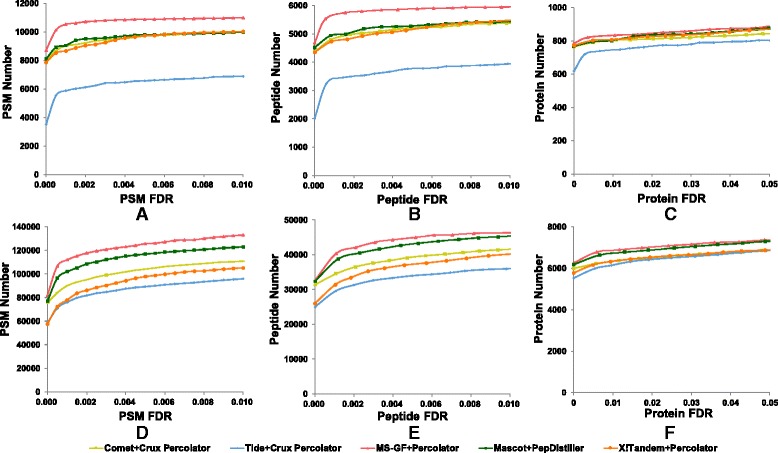
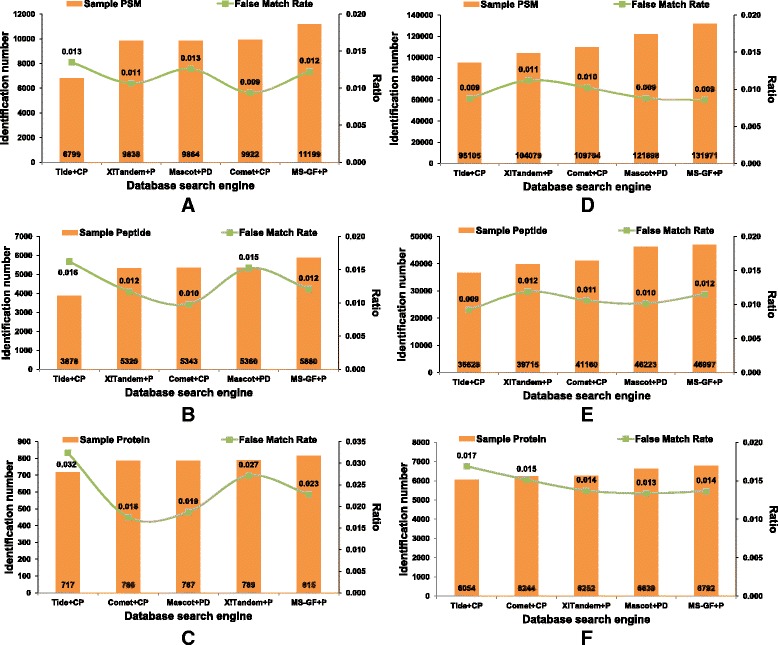
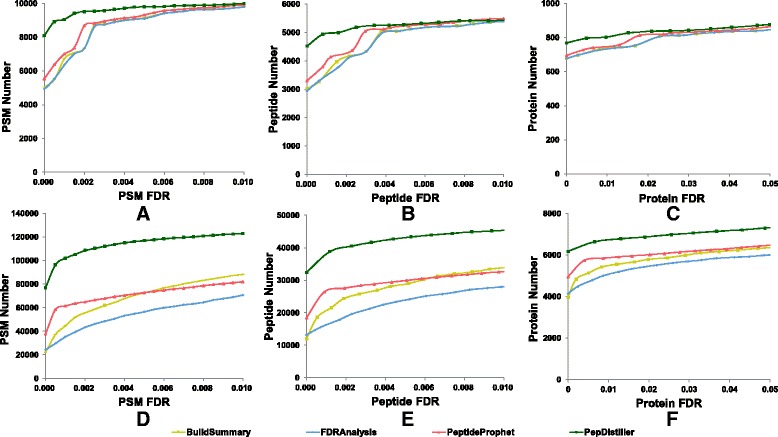
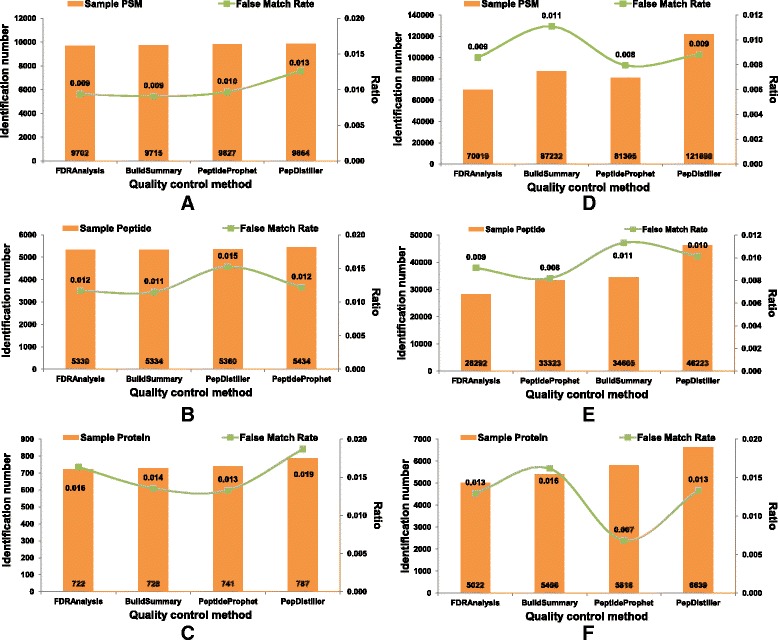
As a supplement to target-decoy strategy, the entrapment sequence method was introduced to assess the key steps of mass spectrometry data analysis process, database search engines and quality control methods. Using the entrapment sequences as the standard, we evaluated five database search engines for both the origanal scores and reprocessed scores, as well as four quality control methods in term of quantity and quality aspects. Our results showed that the latest developed search engine MS-GF+ and percolator-embeded quality control method PepDistiller performed best in all tools respectively. Combined with efficient quality control methods, the search engines can improve the low sensitivity of their original scores. Moreover, based on the entrapment sequence method, we proved that filtering the identifications separately could increase the number of identified peptides while improving the confidence level.
In this study, we have proved that the entrapment sequence method could be an useful strategy to assess the key steps of the mass spectrometry data analysis process. Its applications can be extended to all steps of the common workflow, such as the protein assembling methods and data integration methods.
基于质谱的技术流程为后基因组生物学提供了一个高通量、高灵敏度和高分辨率的平台。不同的工具实现了各种模型和算法来改进蛋白质组学数据分析。目标-诱饵搜索策略已成为控制肽段和蛋白质鉴定中错误识别的最流行策略。虽然该策略可以估计数据集中的错误发现率(FDR),但它不能直接评估目标鉴定中的假阳性匹配。
作为目标-诱饵策略的补充,引入了截留序列方法来评估质谱数据分析过程、数据库搜索引擎和质量控制方法的关键步骤。以截留序列为标准,我们评估了五个数据库搜索引擎的原始分数和重新处理后的分数,以及四种质量控制方法在数量和质量方面的表现。我们的结果表明,最新开发的搜索引擎MS-GF+和嵌入percolator的质量控制方法PepDistiller在所有工具中分别表现最佳。结合高效的质量控制方法,搜索引擎可以提高其原始分数的低灵敏度。此外,基于截留序列方法,我们证明了分别过滤鉴定结果可以增加鉴定出的肽段数量,同时提高置信度。
在本研究中,我们证明了截留序列方法可能是评估质谱数据分析过程关键步骤的一种有用策略。其应用可以扩展到常见工作流程的所有步骤,如蛋白质组装方法和数据整合方法。