Granholm Viktor, Kim Sangtae, Navarro José C F, Sjölund Erik, Smith Richard D, Käll Lukas
Department of Biochemistry and Biophysics, Science for Life Laboratory, Stockholm University , Solna, Sweden.
J Proteome Res. 2014 Feb 7;13(2):890-7. doi: 10.1021/pr400937n. Epub 2013 Dec 23.
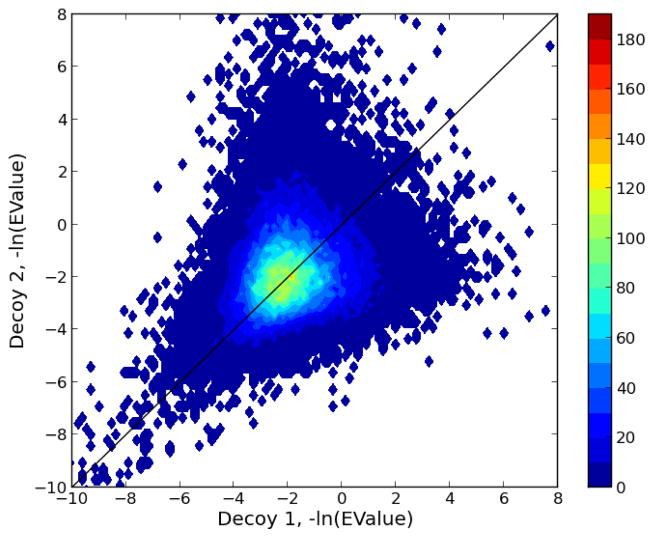
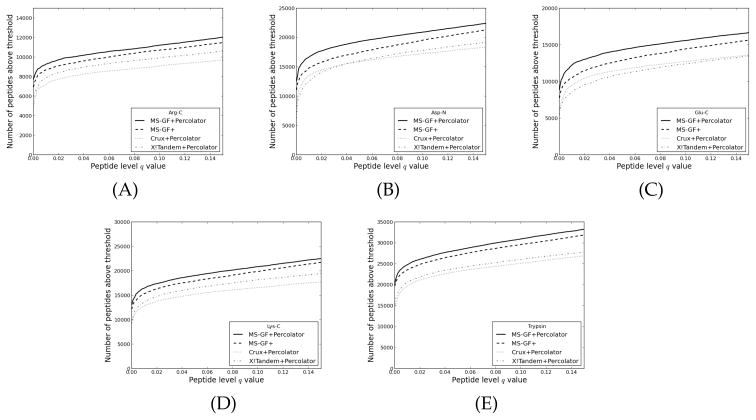
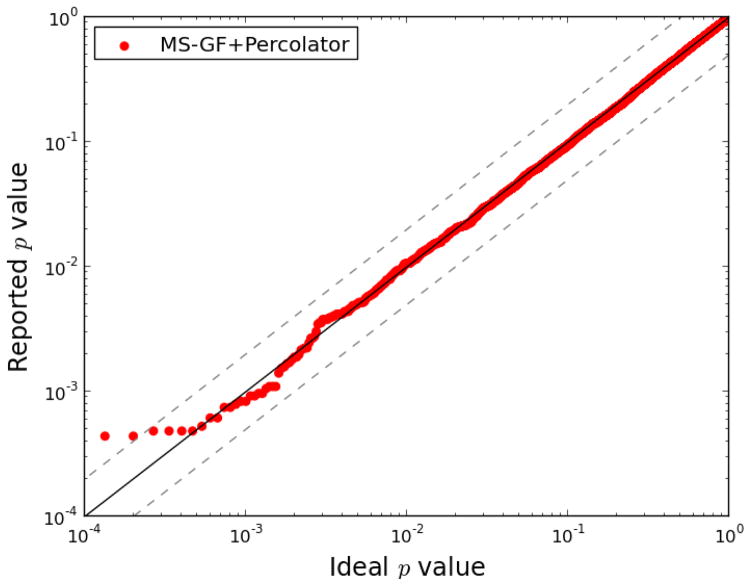
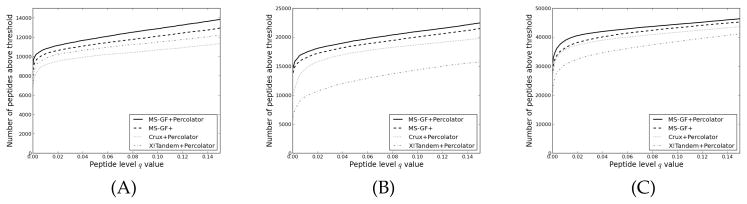
One can interpret fragmentation spectra stemming from peptides in mass-spectrometry-based proteomics experiments using so-called database search engines. Frequently, one also runs post-processors such as Percolator to assess the confidence, infer unique peptides, and increase the number of identifications. A recent search engine, MS-GF+, has shown promising results, due to a new and efficient scoring algorithm. However, MS-GF+ provides few statistical estimates about the peptide-spectrum matches, hence limiting the biological interpretation. Here, we enabled Percolator processing for MS-GF+ output and observed an increased number of identified peptides for a wide variety of data sets. In addition, Percolator directly reports p values and false discovery rate estimates, such as q values and posterior error probabilities, for peptide-spectrum matches, peptides, and proteins, functions that are useful for the whole proteomics community.
在基于质谱的蛋白质组学实验中,可以使用所谓的数据库搜索引擎来解释源自肽段的碎裂谱图。通常,人们还会运行诸如Percolator之类的后处理器来评估可信度、推断独特肽段并增加鉴定数量。最近的一种搜索引擎MS-GF+,由于采用了一种新的高效评分算法,已显示出令人鼓舞的结果。然而,MS-GF+对肽段-谱图匹配提供的统计估计很少,因此限制了生物学解释。在这里,我们对MS-GF+的输出启用了Percolator处理,并观察到在各种数据集上鉴定出的肽段数量有所增加。此外,Percolator直接报告肽段-谱图匹配、肽段和蛋白质的p值以及错误发现率估计值,如q值和后验错误概率,这些功能对整个蛋白质组学领域都很有用。