Weng Shun-Long, Huang Kai-Yao, Kaunang Fergie Joanda, Huang Chien-Hsun, Kao Hui-Ju, Chang Tzu-Hao, Wang Hsin-Yao, Lu Jang-Jih, Lee Tzong-Yi
Department of Obstetrics and Gynecology, Hsinchu Mackay Memorial Hospital, Hsin-Chu, 300, Taiwan.
Mackay Medicine, Nursing and Management College, Taipei, 112, Taiwan.
BMC Bioinformatics. 2017 Mar 14;18(Suppl 3):66. doi: 10.1186/s12859-017-1472-8.
Protein carbonylation, an irreversible and non-enzymatic post-translational modification (PTM), is often used as a marker of oxidative stress. When reactive oxygen species (ROS) oxidized the amino acid side chains, carbonyl (CO) groups are produced especially on Lysine (K), Arginine (R), Threonine (T), and Proline (P). Nevertheless, due to the lack of information about the carbonylated substrate specificity, we were encouraged to develop a systematic method for a comprehensive investigation of protein carbonylation sites.
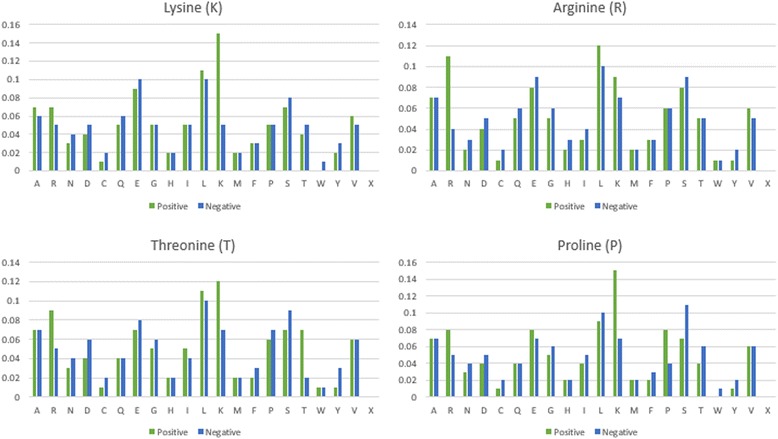
After the removal of redundant data from multipe carbonylation-related articles, totally 226 carbonylated proteins in human are regarded as training dataset, which consisted of 307, 126, 128, and 129 carbonylation sites for K, R, T and P residues, respectively. To identify the useful features in predicting carbonylation sites, the linear amino acid sequence was adopted not only to build up the predictive model from training dataset, but also to compare the effectiveness of prediction with other types of features including amino acid composition (AAC), amino acid pair composition (AAPC), position-specific scoring matrix (PSSM), positional weighted matrix (PWM), solvent-accessible surface area (ASA), and physicochemical properties. The investigation of position-specific amino acid composition revealed that the positively charged amino acids (K and R) are remarkably enriched surrounding the carbonylated sites, which may play a functional role in discriminating between carbonylation and non-carbonylation sites. A variety of predictive models were built using various features and three different machine learning methods. Based on the evaluation by five-fold cross-validation, the models trained with PWM feature could provide better sensitivity in the positive training dataset, while the models trained with AAindex feature achieved higher specificity in the negative training dataset. Additionally, the model trained using hybrid features, including PWM, AAC and AAindex, obtained best MCC values of 0.432, 0.472, 0.443 and 0.467 on K, R, T and P residues, respectively.
When comparing to an existing prediction tool, the selected models trained with hybrid features provided a promising accuracy on an independent testing dataset. In short, this work not only characterized the carbonylated substrate preference, but also demonstrated that the proposed method could provide a feasible means for accelerating preliminary discovery of protein carbonylation.
蛋白质羰基化是一种不可逆的非酶促翻译后修饰(PTM),常被用作氧化应激的标志物。当活性氧(ROS)氧化氨基酸侧链时,尤其是在赖氨酸(K)、精氨酸(R)、苏氨酸(T)和脯氨酸(P)上会产生羰基(CO)基团。然而,由于缺乏关于羰基化底物特异性的信息,我们受到鼓舞去开发一种系统方法来全面研究蛋白质羰基化位点。
从多篇与羰基化相关的文章中去除冗余数据后,总共将人类中的226种羰基化蛋白质视为训练数据集,其中分别包含307、126、128和129个K、R、T和P残基的羰基化位点。为了识别预测羰基化位点的有用特征,不仅采用线性氨基酸序列从训练数据集中构建预测模型,还将其与其他类型的特征进行预测效果比较,这些特征包括氨基酸组成(AAC)、氨基酸对组成(AAPC)、位置特异性评分矩阵(PSSM)、位置加权矩阵(PWM)、溶剂可及表面积(ASA)和物理化学性质。对位置特异性氨基酸组成的研究表明,带正电荷的氨基酸(K和R)在羰基化位点周围显著富集,这可能在区分羰基化和非羰基化位点中发挥功能作用。使用各种特征和三种不同的机器学习方法构建了多种预测模型。基于五折交叉验证的评估,用PWM特征训练的模型在阳性训练数据集中能提供更好的敏感性,而用AAindex特征训练的模型在阴性训练数据集中具有更高的特异性。此外,使用包括PWM、AAC和AAindex在内的混合特征训练的模型在K、R、T和P残基上分别获得了最佳的马修斯相关系数值0.432、0.472、0.443和0.467。
与现有的预测工具相比,选择的用混合特征训练的模型在独立测试数据集上具有可观的准确性。简而言之,这项工作不仅表征了羰基化底物偏好,还证明了所提出的方法可为加速蛋白质羰基化的初步发现提供一种可行的手段。