Raven Kathy E, Gouliouris Theodore, Brodrick Hayley, Coll Francesc, Brown Nicholas M, Reynolds Rosy, Reuter Sandra, Török M Estée, Parkhill Julian, Peacock Sharon J
Department of Medicine, University of Cambridge.
Public Health England, Clinical Microbiology and Public Health Laboratory, Addenbrooke's Hospital, and.
Clin Infect Dis. 2017 Apr 1;64(7):886-893. doi: 10.1093/cid/ciw872.
Vancomycin-resistant Enterococcus faecium (VREfm) is a leading cause of nosocomial infection. Here, we describe the utility of whole-genome sequencing in defining nosocomial VREfm transmission.
A retrospective study at a single hospital in the United Kingdom identified 342 patients with E. faecium bloodstream infection over 7 years. Of these, 293 patients had a stored isolate and formed the basis for the study. The first stored isolate from each case was sequenced (200 VREfm [197 vanA, 2 vanB, and 1 isolate containing both vanA and vanB], 93 vancomycin-susceptible E. faecium) and epidemiological data were collected. Genomes were also available for E. faecium associated with bloodstream infections in 15 patients in neighboring hospitals, and 456 patients across the United Kingdom and Ireland.
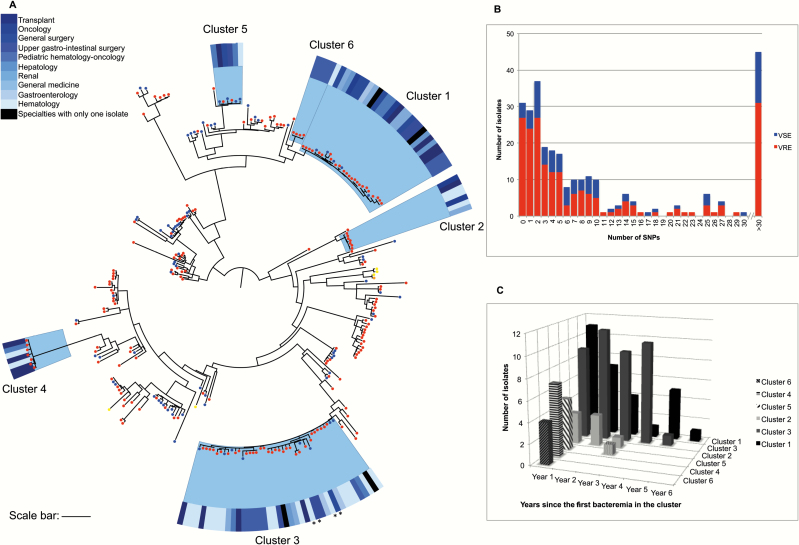
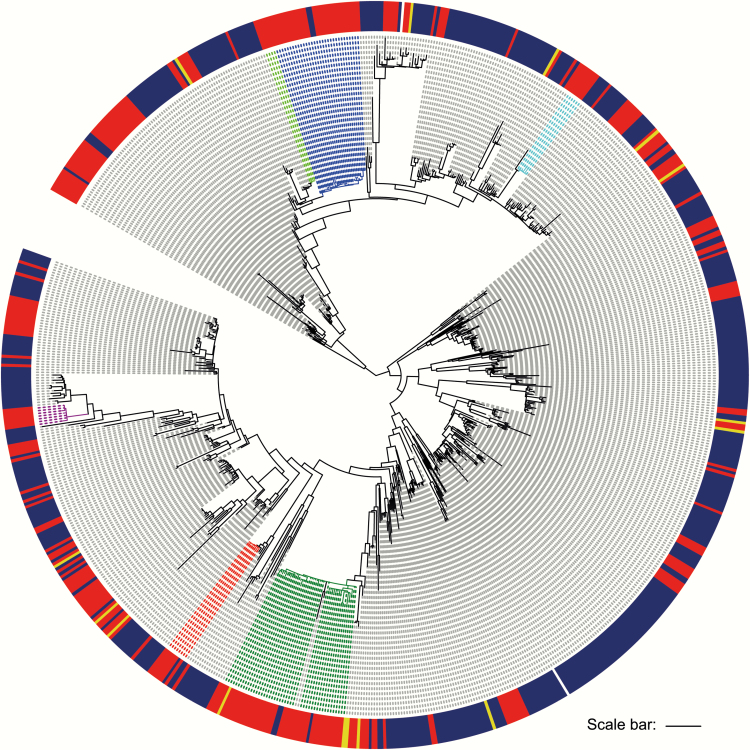
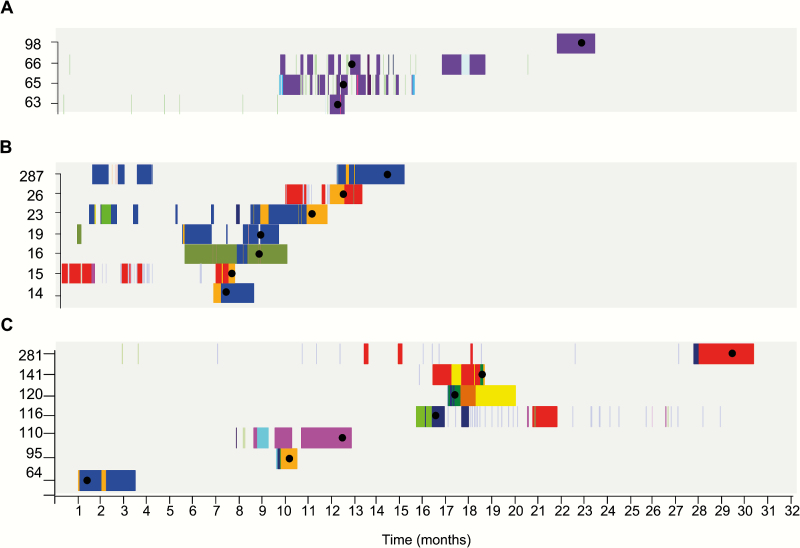
The majority of infections in the 293 patients were hospital-acquired (n = 249) or healthcare-associated (n = 42). Phylogenetic analysis showed that 291 of 293 isolates resided in a hospital-associated clade that contained numerous discrete clusters of closely related isolates, indicative of multiple introductions into the hospital followed by clonal expansion associated with transmission. Fine-scale analysis of 6 exemplar phylogenetic clusters containing isolates from 93 patients (32%) identified complex transmission routes that spanned numerous wards and years, extending beyond the detection of conventional infection control. These contained both vancomycin-resistant and -susceptible isolates. We also identified closely related isolates from patients at Cambridge University Hospitals NHS Foundation Trust and regional and national hospitals, suggesting interhospital transmission.
These findings provide important insights for infection control practice and signpost areas for interventions. We conclude that sequencing represents a powerful tool for the enhanced surveillance and control of nosocomial E. faecium transmission and infection.
耐万古霉素屎肠球菌(VREfm)是医院感染的主要原因。在此,我们描述全基因组测序在确定医院内VREfm传播中的作用。
在英国一家医院进行的一项回顾性研究,确定了7年间342例屎肠球菌血流感染患者。其中,293例患者有保存的分离株,构成了本研究的基础。对每个病例的首个保存分离株进行测序(200株VREfm[197株vanA、2株vanB和1株同时含有vanA和vanB的分离株],93株万古霉素敏感屎肠球菌),并收集流行病学数据。还获得了与邻近医院15例患者以及英国和爱尔兰456例患者血流感染相关的屎肠球菌基因组。
293例患者中的大多数感染为医院获得性(n = 249)或医疗保健相关(n = 42)。系统发育分析表明,293株分离株中的291株属于医院相关分支,其中包含许多紧密相关分离株的离散簇,表明多次引入医院后伴随与传播相关的克隆扩增。对包含93例患者(32%)分离株的6个典型系统发育簇进行的精细分析确定了跨越多个病房和数年的复杂传播途径,超出了传统感染控制的检测范围。这些簇同时包含耐万古霉素和敏感分离株。我们还从剑桥大学医院国民保健服务基金会信托基金以及地区和国家医院的患者中鉴定出密切相关的分离株,提示医院间传播。
这些发现为感染控制实践提供了重要见解,并为干预措施指明了方向。我们得出结论,测序是加强医院内屎肠球菌传播和感染监测与控制的有力工具。