Löytynoja Ari, Goldman Nick
Institute of Biotechnology, University of Helsinki, 00014 Helsinki, Finland.
European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Hinxton, CB10 1SD, United Kingdom.
Genome Res. 2017 Jun;27(6):1039-1049. doi: 10.1101/gr.214973.116. Epub 2017 Apr 6.
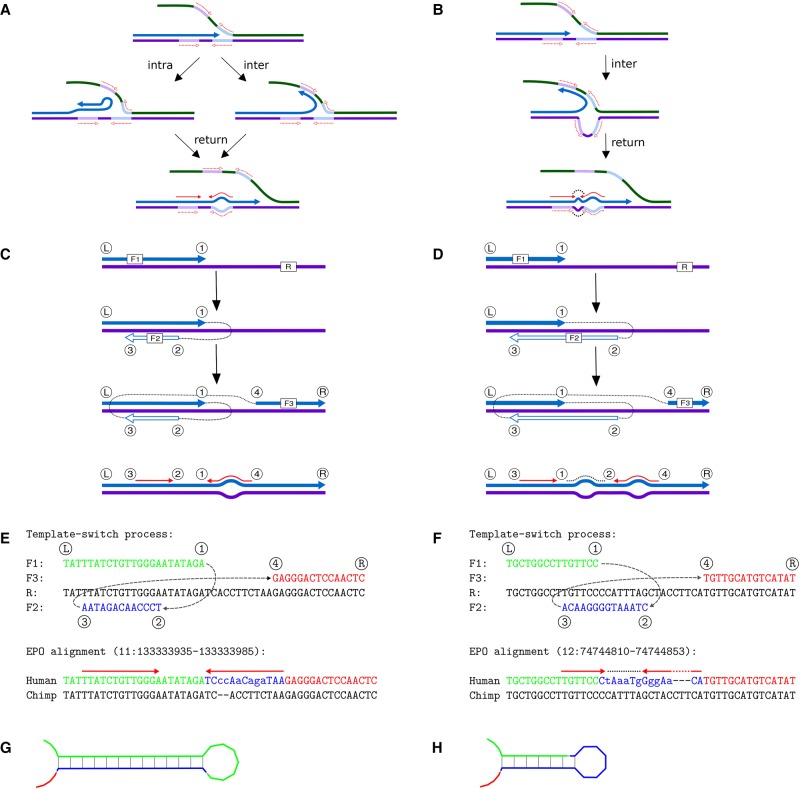
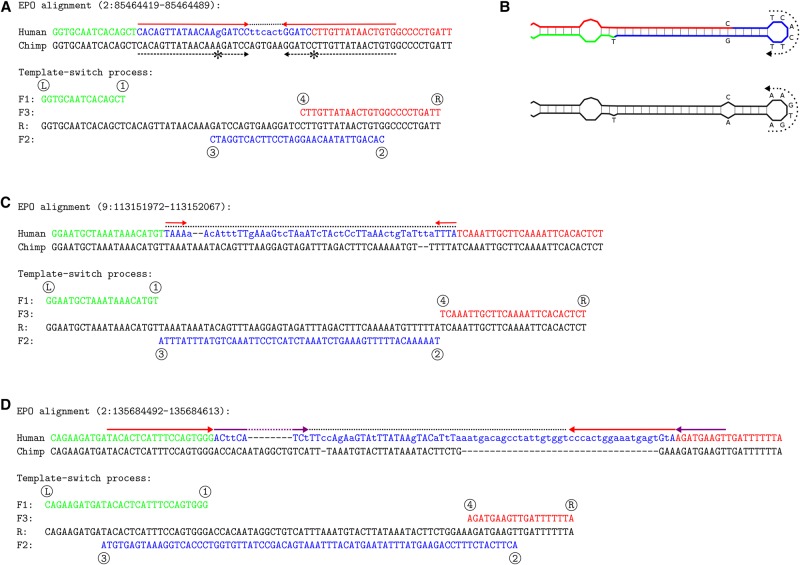
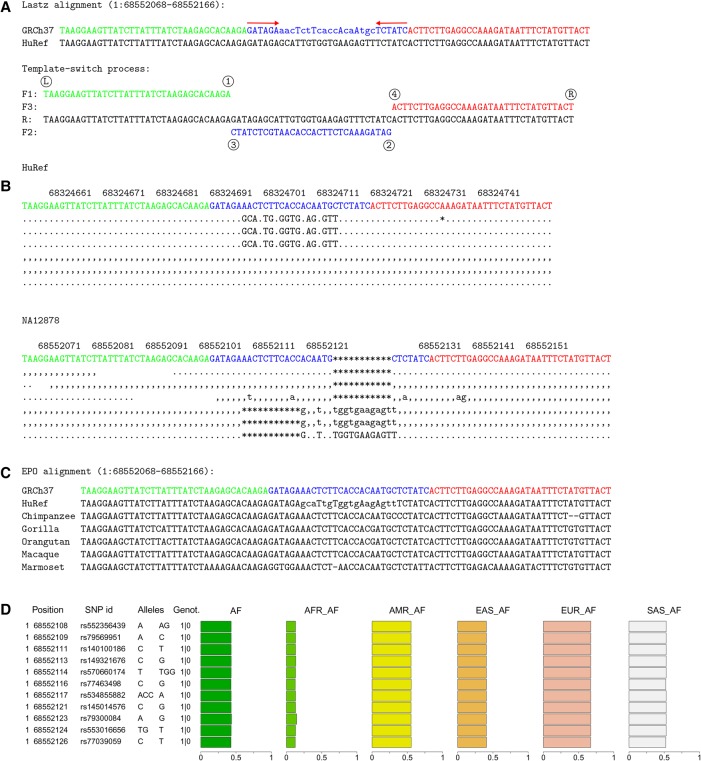
Resequencing efforts are uncovering the extent of genetic variation in humans and provide data to study the evolutionary processes shaping our genome. One recurring puzzle in both intra- and inter-species studies is the high frequency of complex mutations comprising multiple nearby base substitutions or insertion-deletions. We devised a generalized mutation model of template switching during replication that extends existing models of genome rearrangement and used this to study the role of template switch events in the origin of short mutation clusters. Applied to the human genome, our model detects thousands of template switch events during the evolution of human and chimp from their common ancestor and hundreds of events between two independently sequenced human genomes. Although many of these are consistent with a template switch mechanism previously proposed for bacteria, our model also identifies new types of mutations that create short inversions, some flanked by paired inverted repeats. The local template switch process can create numerous complex mutation patterns, including hairpin loop structures, and explains multinucleotide mutations and compensatory substitutions without invoking positive selection, speculative mechanisms, or implausible coincidence. Clustered sequence differences are challenging for current mapping and variant calling methods, and we show that many erroneous variant annotations exist in human reference data. Local template switch events may have been neglected as an explanation for complex mutations because of biases in commonly used analyses. Incorporation of our model into reference-based analysis pipelines and comparisons of de novo assembled genomes will lead to improved understanding of genome variation and evolution.
重测序工作正在揭示人类遗传变异的程度,并提供数据以研究塑造我们基因组的进化过程。种内和种间研究中一个反复出现的谜题是包含多个相邻碱基替换或插入缺失的复杂突变的高频率。我们设计了一种复制过程中模板切换的广义突变模型,该模型扩展了现有的基因组重排模型,并用此来研究模板切换事件在短突变簇起源中的作用。应用于人类基因组时,我们的模型在人类和黑猩猩从共同祖先进化过程中检测到数千次模板切换事件,以及在两个独立测序的人类基因组之间检测到数百次事件。尽管其中许多与先前为细菌提出的模板切换机制一致,但我们的模型还识别出了产生短倒位的新型突变,其中一些两侧是成对的反向重复序列。局部模板切换过程可以产生众多复杂的突变模式,包括发夹环结构,并解释多核苷酸突变和补偿性替换,而无需诉诸正选择、推测机制或难以置信的巧合。成簇的序列差异对当前的定位和变异检测方法具有挑战性,并且我们表明人类参考数据中存在许多错误的变异注释。由于常用分析中的偏差,局部模板切换事件作为复杂突变的一种解释可能被忽视了。将我们的模型纳入基于参考的分析流程以及对从头组装基因组的比较,将有助于更好地理解基因组变异和进化。