Wood Libby, Cordts Isabell, Atalaia Antonio, Marini-Bettolo Chiara, Maddison Paul, Phillips Margaret, Roberts Mark, Rogers Mark, Hammans Simon, Straub Volker, Petty Richard, Orrell Richard, Monckton Darren G, Nikolenko Nikoletta, Jimenez-Moreno Aura Cecilia, Thompson Rachel, Hilton-Jones David, Turner Chris, Lochmüller Hanns
John Walton Muscular Dystrophy Research Centre, Institute of Genetic Medicine, Newcastle University, Newcastle upon Tyne, UK.
Department of Neurology, RWTH Aachen University, Aachen, Germany.
J Neurol. 2017 May;264(5):979-988. doi: 10.1007/s00415-017-8483-2. Epub 2017 Apr 10.
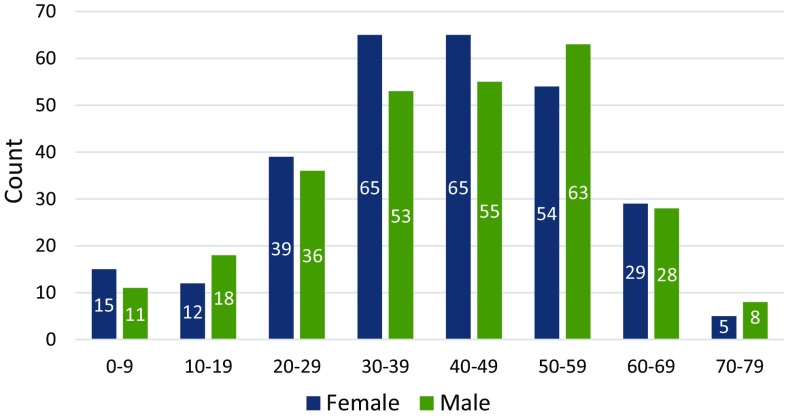
Myotonic dystrophy type 1 (DM1) is the most frequent muscular dystrophy worldwide with complex, multi-systemic, and progressively worsening symptoms. There is currently no treatment for this inherited disorder and research can be challenging due to the rarity and variability of the disease. The UK Myotonic Dystrophy Patient Registry is a patient self-enrolling online database collecting clinical and genetic information. For this cross-sectional "snapshot" analysis, 556 patients with a confirmed diagnosis of DM1 registered between May 2012 and July 2016 were included. An almost even distribution was seen between genders and a broad range of ages was present from 8 months to 78 years, with the largest proportion between 30 and 59 years. The two most frequent symptoms were fatigue and myotonia, reported by 79 and 78% of patients, respectively. The severity of myotonia correlated with the severity of fatigue as well as mobility impairment, and dysphagia occurred mostly in patients also reporting myotonia. Men reported significantly more frequent severe myotonia, whereas severe fatigue was more frequently reported by women. Cardiac abnormalities were diagnosed in 48% of patients and more than one-third of them needed a cardiac implant. Fifteen percent of patients used a non-invasive ventilation and cataracts were removed in 26% of patients, 65% of which before the age of 50 years. The registry's primary aim was to facilitate and accelerate clinical research. However, these data also allow us to formulate questions for hypothesis-driven research that may lead to improvements in care and treatment.
1型强直性肌营养不良(DM1)是全球最常见的肌营养不良症,症状复杂、多系统且逐渐恶化。目前,这种遗传性疾病尚无治疗方法,由于该疾病的罕见性和变异性,相关研究颇具挑战性。英国强直性肌营养不良患者登记处是一个患者可自行在线注册的数据库,用于收集临床和基因信息。在这项横断面“快照”分析中,纳入了2012年5月至2016年7月期间确诊为DM1并注册的556例患者。性别分布几乎均匀,年龄范围从8个月至78岁,其中最大比例在30至59岁之间。最常见的两种症状是疲劳和肌强直,分别有79%和78%的患者报告出现。肌强直的严重程度与疲劳及活动障碍的严重程度相关,吞咽困难主要发生在也报告有肌强直的患者中。男性报告严重肌强直的频率显著更高,而女性报告严重疲劳的频率更高。48%的患者被诊断出心脏异常,其中超过三分之一的患者需要心脏植入装置。15%的患者使用无创通气,26%的患者摘除了白内障,其中65%在50岁之前。该登记处的主要目标是促进和加速临床研究。然而,这些数据也使我们能够提出一些问题,用于假设驱动的研究,这可能会改善护理和治疗。