Huang Shengfeng, Kang Mingjing, Xu Anlong
State Key Laboratory of Biocontrol, Guangdong Key Laboratory of Pharmaceutical Functional Genes, School of Life Sciences, Sun Yat-Sen University, Guangzhou 510275, People's Republic of China.
Bioinformatics. 2017 Aug 15;33(16):2577-2579. doi: 10.1093/bioinformatics/btx220.
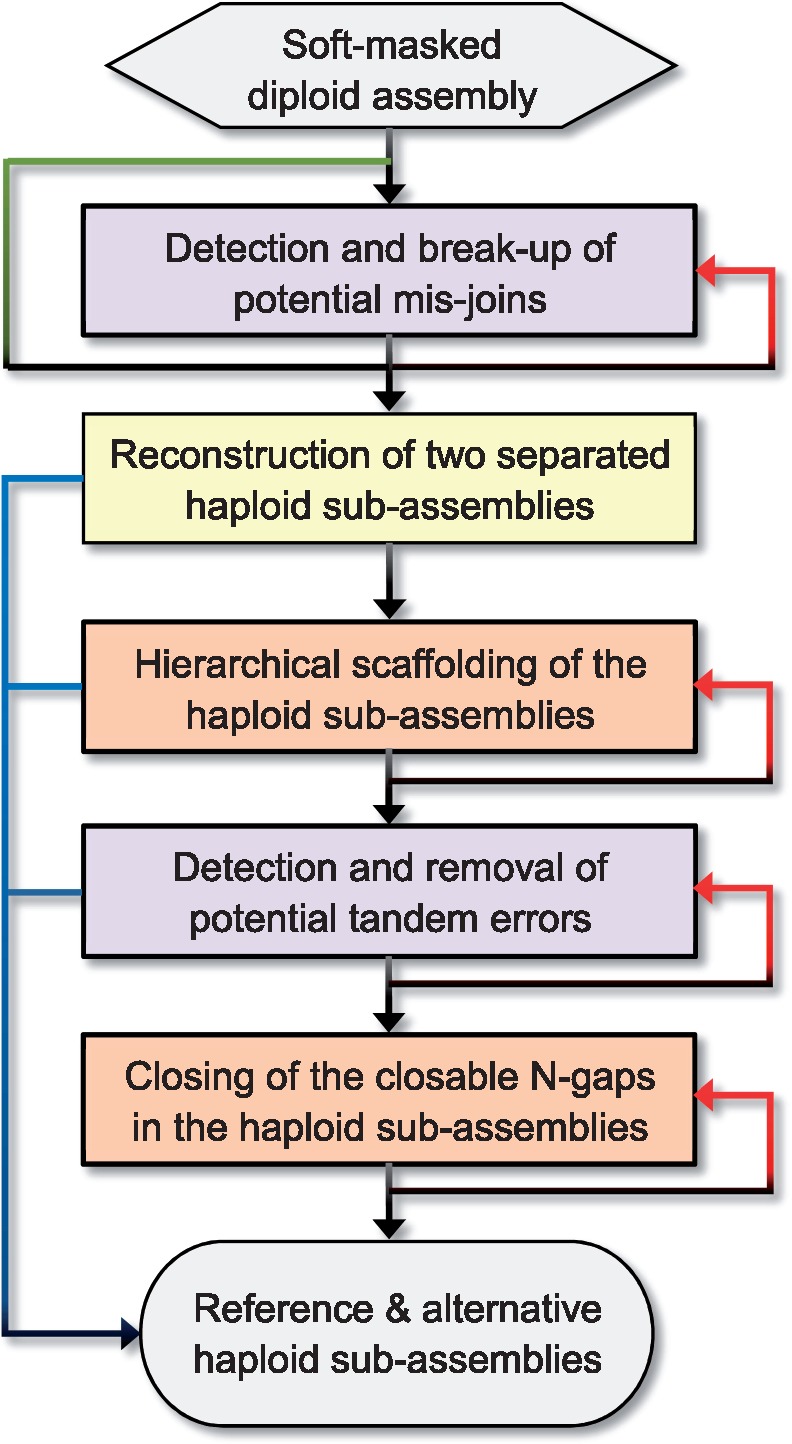
De novo assembly is a difficult issue for heterozygous diploid genomes. The advent of high-throughput short-read and long-read sequencing technologies provides both new challenges and potential solutions to the issue. Here, we present HaploMerger2 (HM2), an automated pipeline for rebuilding both haploid sub-assemblies from the polymorphic diploid genome assembly. It is designed to work on pre-existing diploid assemblies, which are typically created by using de novo assemblers. HM2 can process any diploid assemblies, but it is especially suitable for diploid assemblies with high heterozygosity (≥3%), which can be difficult for other tools. This pipeline also implements flexible and sensitive assembly error detection, a hierarchical scaffolding procedure and a reliable gap-closing method for haploid sub-assemblies. Using HM2, we demonstrate that two haploid sub-assemblies reconstructed from a real, highly-polymorphic diploid assembly show greatly improved continuity.
Source code, executables and the testing dataset are freely available at https://github.com/mapleforest/HaploMerger2/releases/.
Supplementary data are available at Bioinformatics online.
对于杂合二倍体基因组而言,从头组装是一个难题。高通量短读长和长读长测序技术的出现,给这个问题带来了新的挑战,也提供了潜在的解决方案。在此,我们展示了HaploMerger2(HM2),这是一种用于从多态性二倍体基因组组装中重建单倍体子组装的自动化流程。它旨在处理预先存在的二倍体组装,这些组装通常是使用从头组装器创建的。HM2可以处理任何二倍体组装,但它特别适用于高杂合度(≥3%)的二倍体组装,而其他工具处理这类组装可能会有困难。该流程还为单倍体子组装实现了灵活且灵敏的组装错误检测、分层支架构建程序和可靠的缺口闭合方法。使用HM2,我们证明了从一个真实的、高度多态的二倍体组装中重建的两个单倍体子组装具有显著改善的连续性。
源代码、可执行文件和测试数据集可在https://github.com/mapleforest/HaploMerger2/releases/免费获取。
补充数据可在《生物信息学》在线获取。