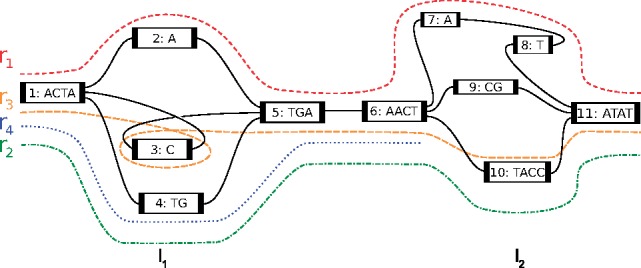
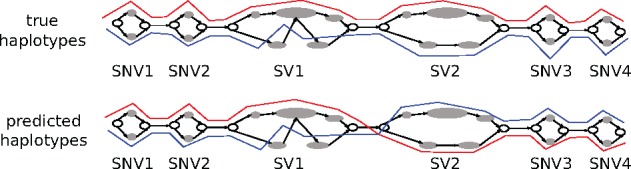
Center for Bioinformatics, Saarland University, Saarland Informatics Campus E2.1, Saarbrücken, Germany.
Department of Computational Biology & Applied Algorithmics, Max Planck Institute for Informatics, Saarland Informatics Campus E1.4, Saarbrücken, Germany.
Bioinformatics. 2018 Jul 1;34(13):i105-i114. doi: 10.1093/bioinformatics/bty279.
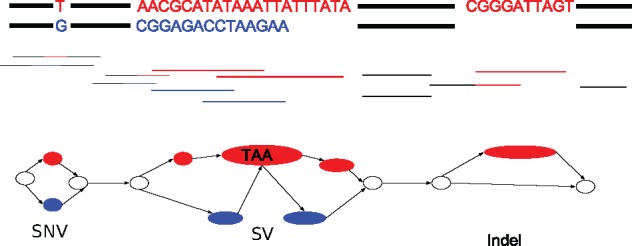
Constructing high-quality haplotype-resolved de novo assemblies of diploid genomes is important for revealing the full extent of structural variation and its role in health and disease. Current assembly approaches often collapse the two sequences into one haploid consensus sequence and, therefore, fail to capture the diploid nature of the organism under study. Thus, building an assembler capable of producing accurate and complete diploid assemblies, while being resource-efficient with respect to sequencing costs, is a key challenge to be addressed by the bioinformatics community.
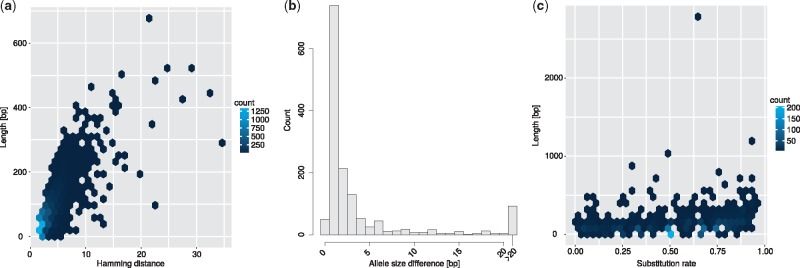
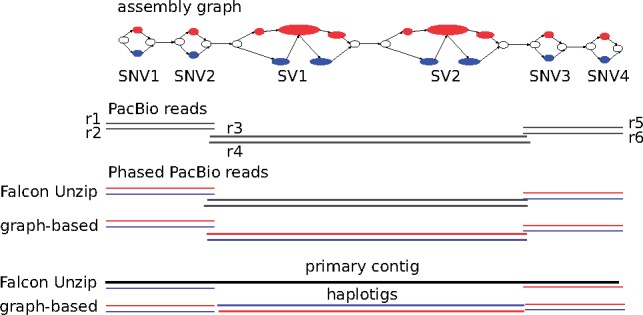
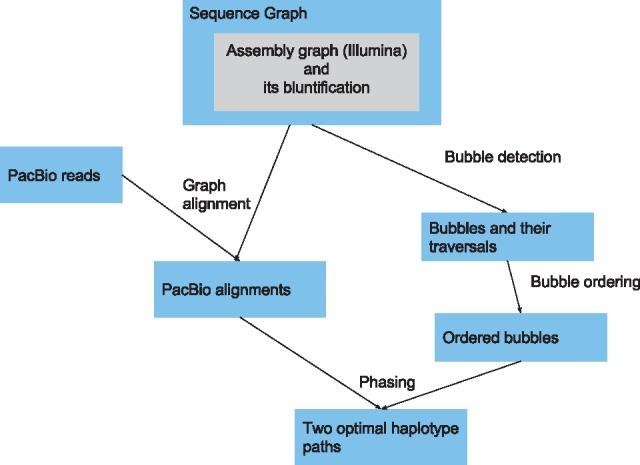
We present a novel graph-based approach to diploid assembly, which combines accurate Illumina data and long-read Pacific Biosciences (PacBio) data. We demonstrate the effectiveness of our method on a pseudo-diploid yeast genome and show that we require as little as 50× coverage Illumina data and 10× PacBio data to generate accurate and complete assemblies. Additionally, we show that our approach has the ability to detect and phase structural variants.
https://github.com/whatshap/whatshap.
Supplementary data are available at Bioinformatics online.
构建高质量的单倍型解析从头组装的二倍体基因组对于揭示结构变异的全部范围及其在健康和疾病中的作用非常重要。目前的组装方法通常将两个序列折叠成一个单倍体共识序列,因此无法捕获研究中生物体的二倍体性质。因此,构建一个能够生成准确完整的二倍体组装的组装器,同时在测序成本方面具有资源效率,是生物信息学社区需要解决的关键挑战。
我们提出了一种新的基于图的二倍体组装方法,该方法结合了准确的 Illumina 数据和长读长 Pacific Biosciences(PacBio)数据。我们在一个拟二倍体酵母基因组上证明了我们方法的有效性,并表明我们只需要 50×Illumina 数据覆盖度和 10×PacBio 数据即可生成准确完整的组装。此外,我们还表明,我们的方法具有检测和相位结构变异的能力。
https://github.com/whatshap/whatshap。
补充数据可在Bioinformatics 在线获得。