Zhang Fantao, Zhou Yi, Zhang Meng, Luo Xiangdong, Xie Jiankun
Plant Functional Genomics Laboratory, College of Life Sciences, Jiangxi Normal University, Nanchang, 330022, China
Plant Functional Genomics Laboratory, College of Life Sciences, Jiangxi Normal University, Nanchang, 330022, China.
Biosci Rep. 2017 Jun 27;37(3). doi: 10.1042/BSR20160509. Print 2017 Jun 30.
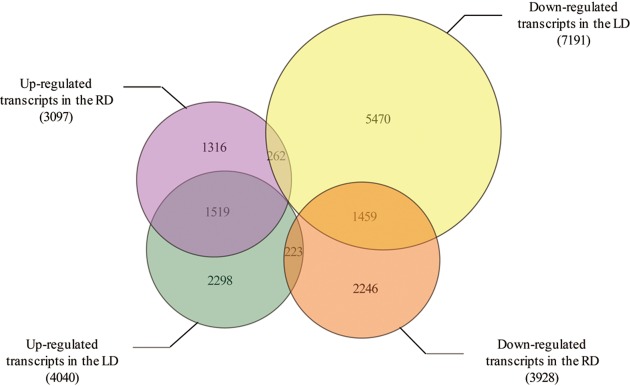
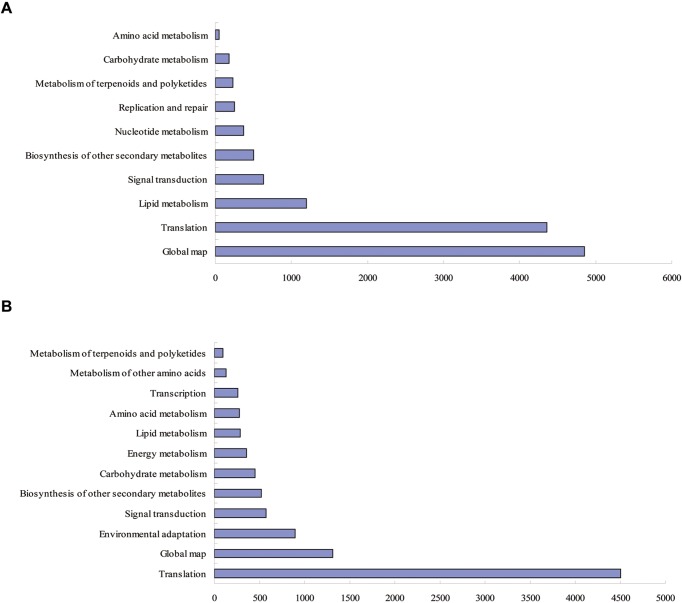
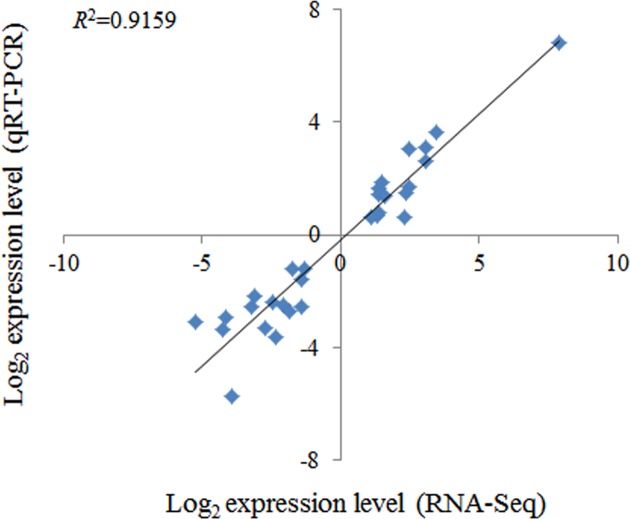
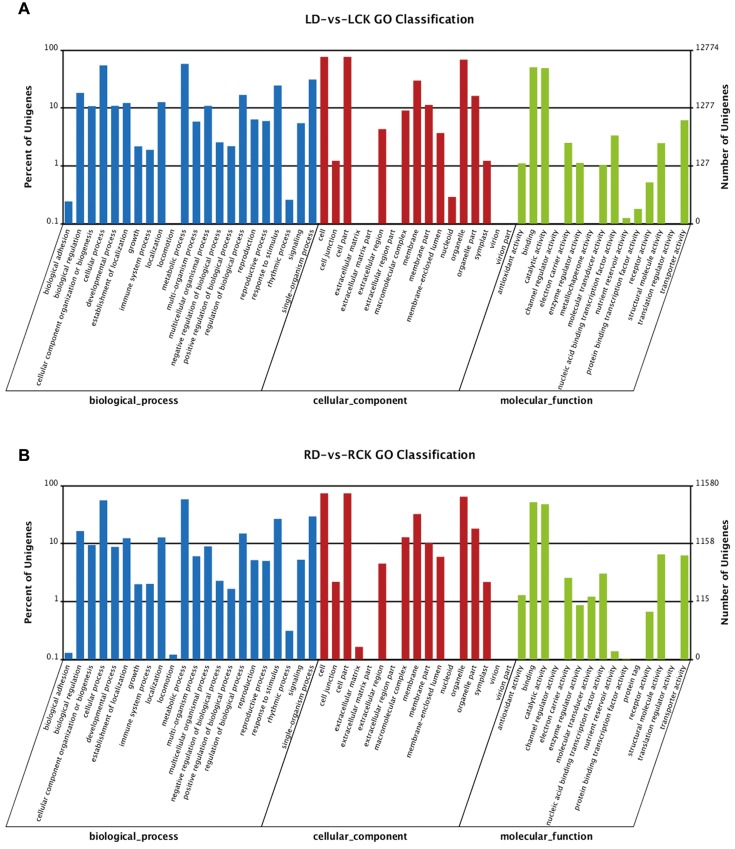
Drought is a serious constraint to rice production throughout the world, and although Dongxiang wild rice (, DXWR) possesses a high degree of drought resistance, the underlying mechanisms of this trait remains unclear. In the present study, cDNA libraries were constructed from the leaf and root tissues of drought-stressed and untreated DXWR seedlings, and transcriptome sequencing was performed with the goal of elucidating the molecular mechanisms involved in drought-stress response. The results indicated that 11231 transcripts were differentially expressed in the leaves (4040 up-regulated and 7191 down-regulated) and 7025 transcripts were differentially expressed in the roots (3097 up-regulated and 3928 down-regulated). Among these differentially expressed genes (DEGs), the detection of many transcriptional factors and functional genes demonstrated that multiple regulatory pathways were involved in drought resistance. Meanwhile, the DEGs were also annotated with gene ontology (GO) terms and key pathways via functional classification and Kyoto Encyclopedia of Gene and Genomes (KEGG) pathway mapping, respectively. A set of the most interesting candidate genes was then identified by combining the DEGs with previously identified drought-resistant quantitative trait loci (QTL). The present work provides abundant genomic information for functional dissection of the drought resistance of DXWR, and findings will further help the current understanding of the biological regulatory mechanisms of drought resistance in plants and facilitate the breeding of new drought-resistant rice cultivars.
干旱是全球水稻生产的严重制约因素,尽管东乡野生稻(DXWR)具有高度的抗旱性,但其抗旱的潜在机制仍不清楚。在本研究中,从干旱胁迫和未处理的DXWR幼苗的叶和根组织构建了cDNA文库,并进行了转录组测序,目的是阐明参与干旱胁迫响应的分子机制。结果表明,11231个转录本在叶片中差异表达(4040个上调和7191个下调),7025个转录本在根中差异表达(3097个上调和3928个下调)。在这些差异表达基因(DEG)中,许多转录因子和功能基因的检测表明多种调控途径参与了抗旱性。同时,通过功能分类和京都基因与基因组百科全书(KEGG)途径映射,分别用基因本体(GO)术语和关键途径对DEG进行了注释。然后通过将DEG与先前鉴定的抗旱数量性状位点(QTL)相结合,鉴定出一组最有趣的候选基因。本研究为东乡野生稻抗旱性的功能解析提供了丰富的基因组信息,研究结果将有助于进一步了解植物抗旱的生物调控机制,并促进新的抗旱水稻品种的培育。