Yin Changchuan, Yau Stephen S-T
Department of Mathematics, Statistics and Computer Science, The University of Illinois at Chicago, Chicago, IL 60607-7045, United States of America.
Department of Mathematical Sciences, Tsinghua University, Beijing 100084, China.
PLoS One. 2017 Apr 21;12(4):e0174862. doi: 10.1371/journal.pone.0174862. eCollection 2017.
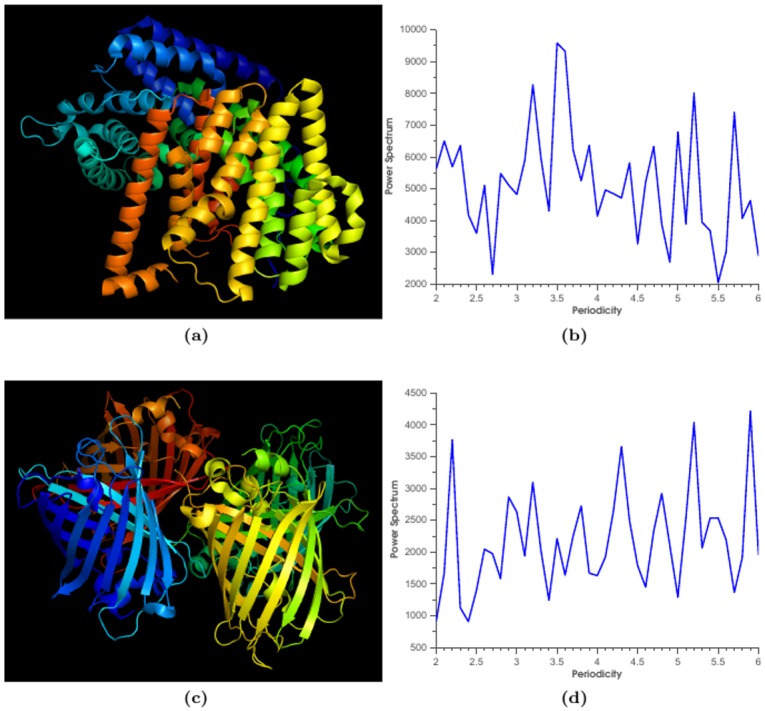
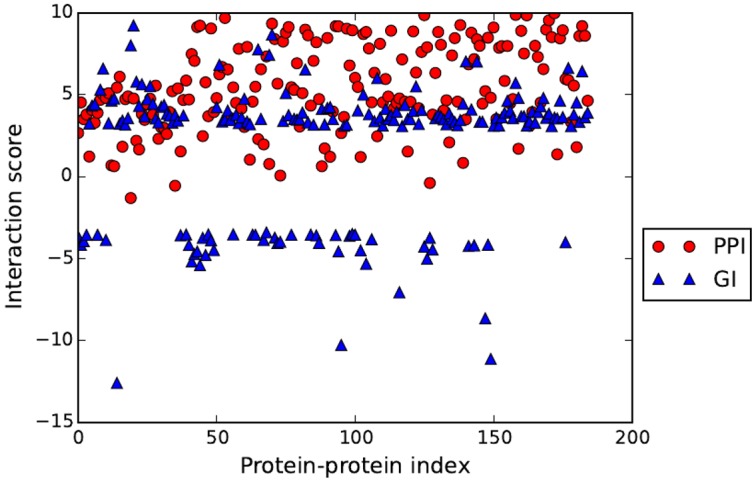
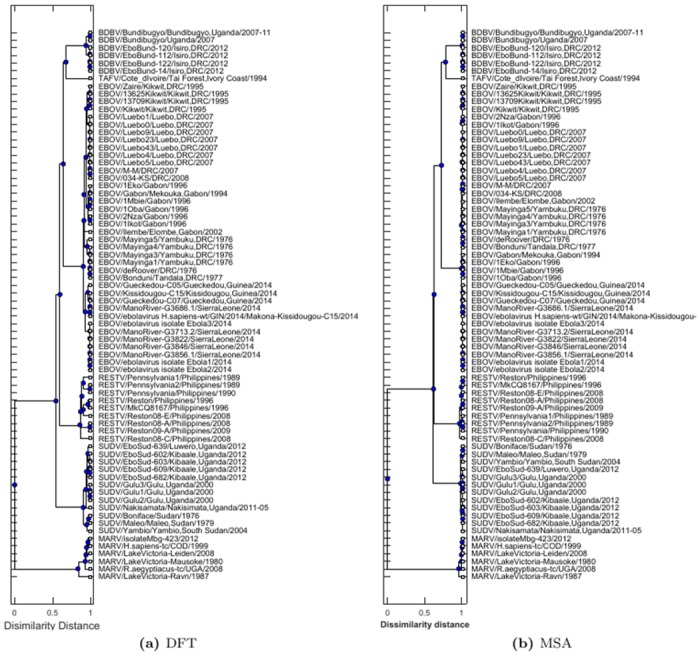
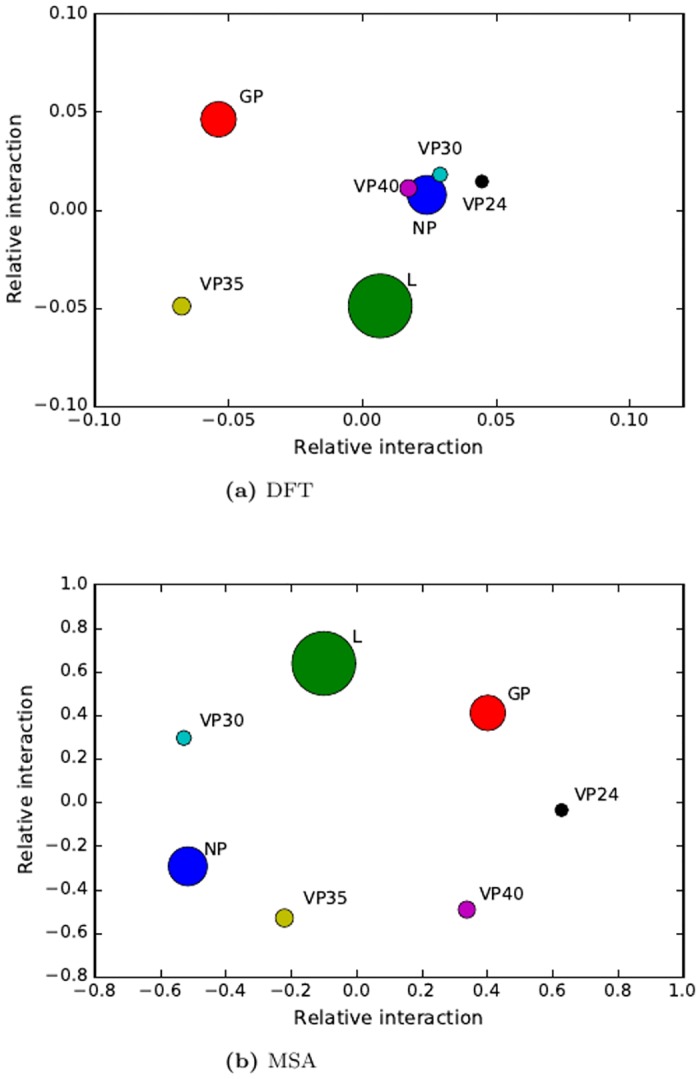
Protein-protein interactions (PPIs) play key roles in life processes, such as signal transduction, transcription regulations, and immune response, etc. Identification of PPIs enables better understanding of the functional networks within a cell. Common experimental methods for identifying PPIs are time consuming and expensive. However, recent developments in computational approaches for inferring PPIs from protein sequences based on coevolution theory avoid these problems. In the coevolution theory model, interacted proteins may show coevolutionary mutations and have similar phylogenetic trees. The existing coevolution methods depend on multiple sequence alignments (MSA); however, the MSA-based coevolution methods often produce high false positive interactions. In this paper, we present a computational method using an alignment-free approach to accurately detect PPIs and reduce false positives. In the method, protein sequences are numerically represented by biochemical properties of amino acids, which reflect the structural and functional differences of proteins. Fourier transform is applied to the numerical representation of protein sequences to capture the dissimilarities of protein sequences in biophysical context. The method is assessed for predicting PPIs in Ebola virus. The results indicate strong coevolution between the protein pairs (NP-VP24, NP-VP30, NP-VP40, VP24-VP30, VP24-VP40, and VP30-VP40). The method is also validated for PPIs in influenza and E.coli genomes. Since our method can reduce false positive and increase the specificity of PPI prediction, it offers an effective tool to understand mechanisms of disease pathogens and find potential targets for drug design. The Python programs in this study are available to public at URL (https://github.com/cyinbox/PPI).
蛋白质-蛋白质相互作用(PPIs)在生命过程中发挥着关键作用,如信号转导、转录调控和免疫反应等。蛋白质-蛋白质相互作用的鉴定有助于更好地理解细胞内的功能网络。鉴定蛋白质-蛋白质相互作用的常见实验方法既耗时又昂贵。然而,最近基于协同进化理论从蛋白质序列推断蛋白质-蛋白质相互作用的计算方法的发展避免了这些问题。在协同进化理论模型中,相互作用的蛋白质可能会显示协同进化突变并具有相似的系统发育树。现有的协同进化方法依赖于多序列比对(MSA);然而,基于多序列比对的协同进化方法往往会产生较高的假阳性相互作用。在本文中,我们提出了一种使用无比对方法来准确检测蛋白质-蛋白质相互作用并减少假阳性的计算方法。在该方法中,蛋白质序列由氨基酸的生化特性进行数值表示,这反映了蛋白质的结构和功能差异。将傅里叶变换应用于蛋白质序列的数值表示,以捕捉生物物理背景下蛋白质序列的差异。该方法在埃博拉病毒中用于预测蛋白质-蛋白质相互作用进行了评估。结果表明蛋白质对(NP-VP24、NP-VP30、NP-VP40、VP24-VP30、VP24-VP40和VP30-VP40)之间存在强烈的协同进化。该方法也在流感和大肠杆菌基因组中的蛋白质-蛋白质相互作用方面得到了验证。由于我们的方法可以减少假阳性并提高蛋白质-蛋白质相互作用预测的特异性,它为理解疾病病原体的机制和寻找药物设计的潜在靶点提供了一个有效的工具。本研究中的Python程序可在URL(https://github.com/cyinbox/PPI)上向公众提供。