Department of Neurosciences, University of New Mexico School of Medicine, Albuquerque, NM, USA.
Department of Brain and Cognitive Sciences, Picower Institute for Learning and Memory, Massachusetts Institute of Technology, Cambridge, MA, USA.
Mol Psychiatry. 2018 Apr;23(4):1051-1065. doi: 10.1038/mp.2017.86. Epub 2017 Apr 25.
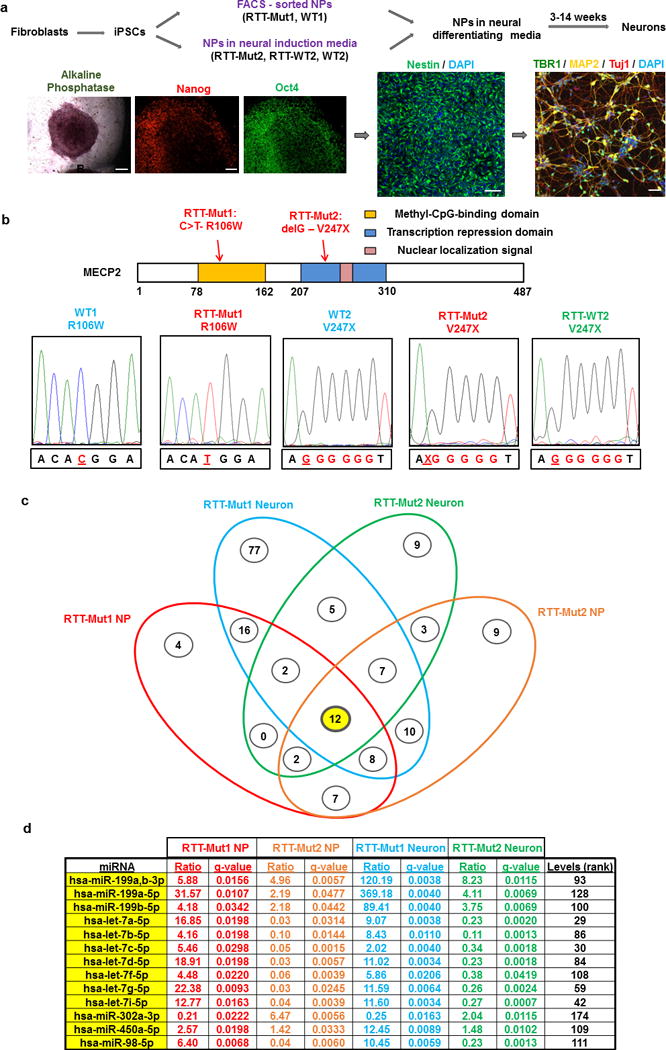
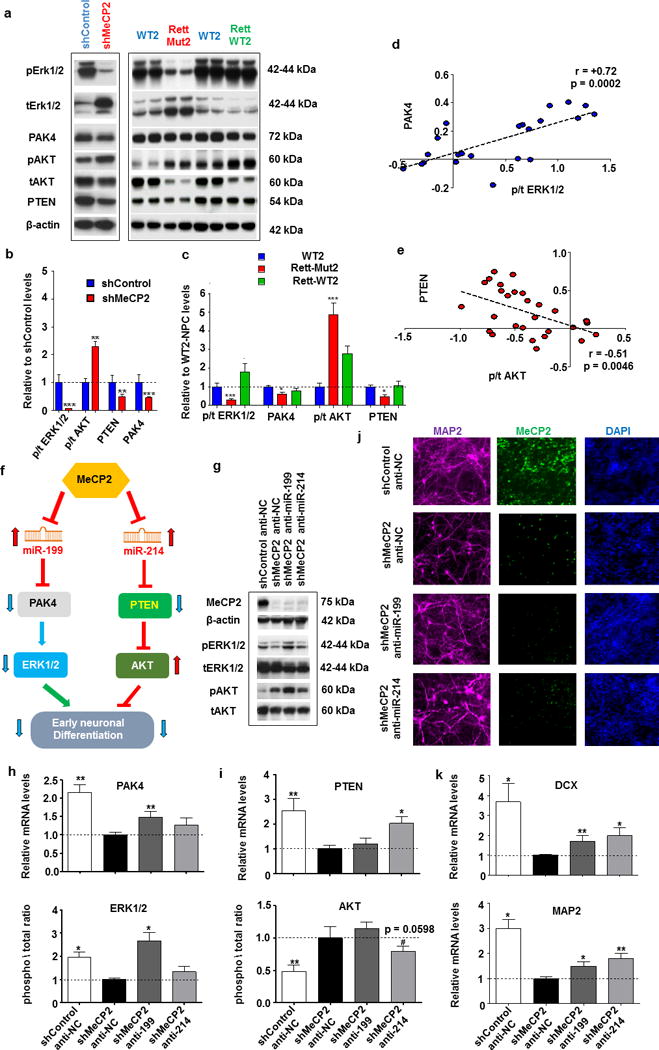
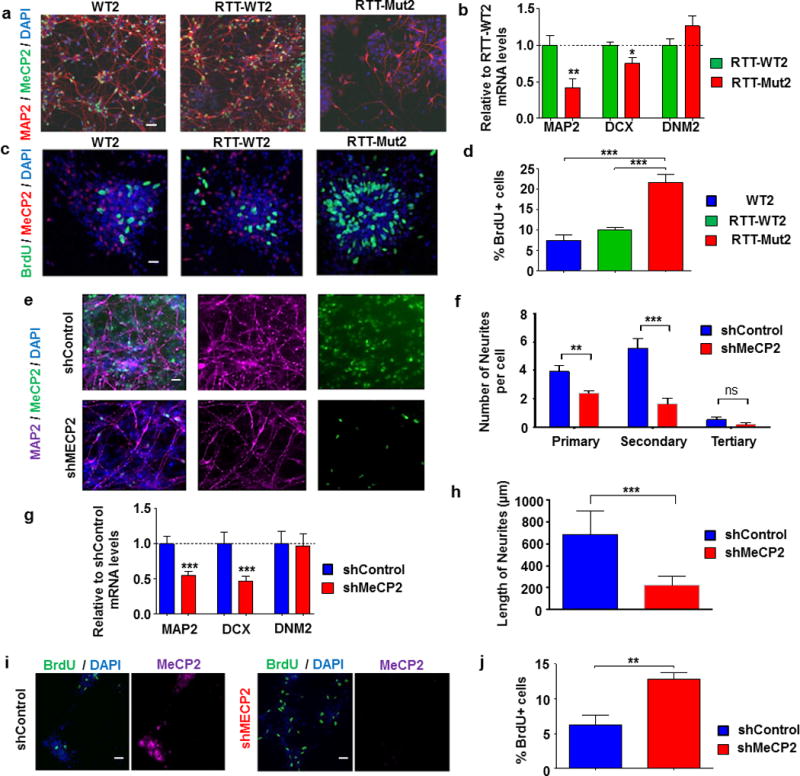
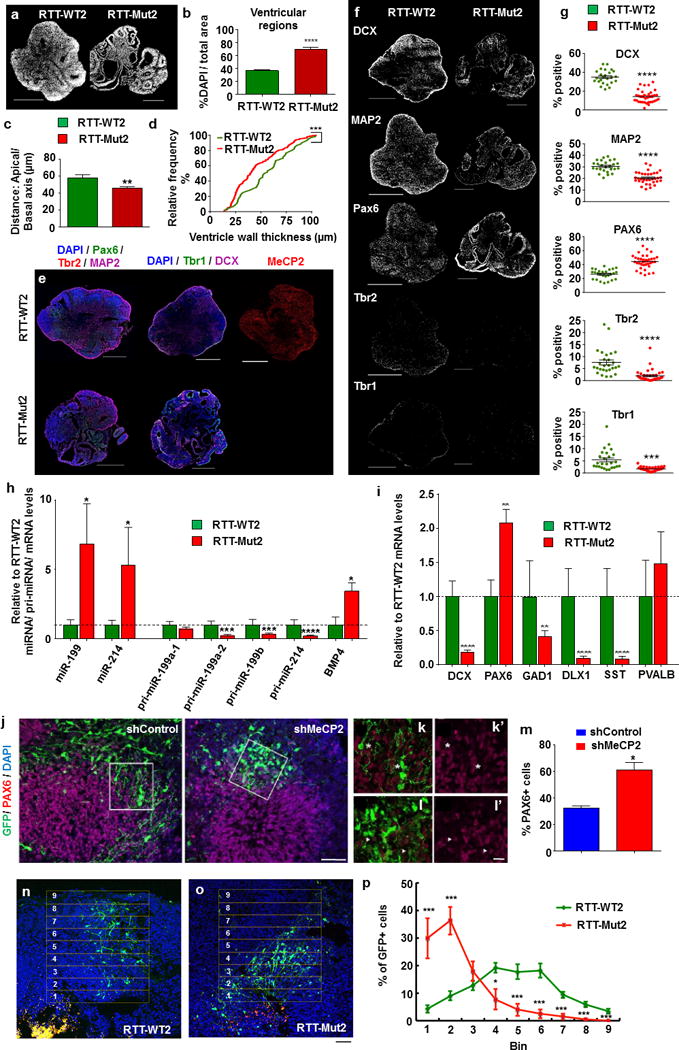
Rett syndrome (RTT) is an X-linked, neurodevelopmental disorder caused primarily by mutations in the methyl-CpG-binding protein 2 (MECP2) gene, which encodes a multifunctional epigenetic regulator with known links to a wide spectrum of neuropsychiatric disorders. Although postnatal functions of MeCP2 have been thoroughly investigated, its role in prenatal brain development remains poorly understood. Given the well-established importance of microRNAs (miRNAs) in neurogenesis, we employed isogenic human RTT patient-derived induced pluripotent stem cell (iPSC) and MeCP2 short hairpin RNA knockdown approaches to identify novel MeCP2-regulated miRNAs enriched during early human neuronal development. Focusing on the most dysregulated miRNAs, we found miR-199 and miR-214 to be increased during early brain development and to differentially regulate extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase and protein kinase B (PKB/AKT) signaling. In parallel, we characterized the effects on human neurogenesis and neuronal differentiation brought about by MeCP2 deficiency using both monolayer and three-dimensional (cerebral organoid) patient-derived and MeCP2-deficient neuronal culture models. Inhibiting miR-199 or miR-214 expression in iPSC-derived neural progenitors deficient in MeCP2 restored AKT and ERK activation, respectively, and ameliorated the observed alterations in neuronal differentiation. Moreover, overexpression of miR-199 or miR-214 in the wild-type mouse embryonic brains was sufficient to disturb neurogenesis and neuronal migration in a similar manner to Mecp2 knockdown. Taken together, our data support a novel miRNA-mediated pathway downstream of MeCP2 that influences neurogenesis via interactions with central molecular hubs linked to autism spectrum disorders.
雷特综合征(RTT)是一种主要由甲基化CpG 结合蛋白 2(MECP2)基因突变引起的 X 连锁神经发育障碍,该基因编码一种多功能表观遗传调节剂,与广泛的神经精神疾病有已知的联系。尽管 MeCP2 的产后功能已被彻底研究,但它在产前大脑发育中的作用仍知之甚少。鉴于 microRNAs(miRNAs)在神经发生中的重要性,我们采用同基因人 RTT 患者衍生的诱导多能干细胞(iPSC)和 MeCP2 短发夹 RNA 敲低方法,鉴定在早期人类神经元发育过程中富集的新型 MeCP2 调节的 miRNAs。关注最失调的 miRNAs,我们发现 miR-199 和 miR-214 在大脑早期发育过程中增加,并差异调节细胞外信号调节激酶(ERK)/丝裂原活化蛋白激酶和蛋白激酶 B(PKB/AKT)信号。同时,我们使用单层和三维(脑类器官)患者衍生和 MeCP2 缺陷神经元培养模型,表征 MeCP2 缺陷对人类神经发生和神经元分化的影响。在 MeCP2 缺陷的 iPSC 衍生神经祖细胞中抑制 miR-199 或 miR-214 的表达分别恢复了 AKT 和 ERK 的激活,并改善了观察到的神经元分化改变。此外,在野生型小鼠胚胎脑中过表达 miR-199 或 miR-214 足以以类似于 Mecp2 敲低的方式干扰神经发生和神经元迁移。总之,我们的数据支持 MeCP2 下游的新型 miRNA 介导途径,该途径通过与自闭症谱系障碍相关的中枢分子枢纽相互作用影响神经发生。