Matsui Yusuke, Niida Atsushi, Uchi Ryutaro, Mimori Koshi, Miyano Satoru, Shimamura Teppei
Division of Systems Biology, Nagoya University Graduate School of Medicine, Nagoya, Japan.
Division of Health Medical Computational Science, Health Intelligence Center, The Institute of Medical Science, The University of Tokyo, Tokyo, Japan.
PLoS Comput Biol. 2017 May 1;13(5):e1005509. doi: 10.1371/journal.pcbi.1005509. eCollection 2017 May.
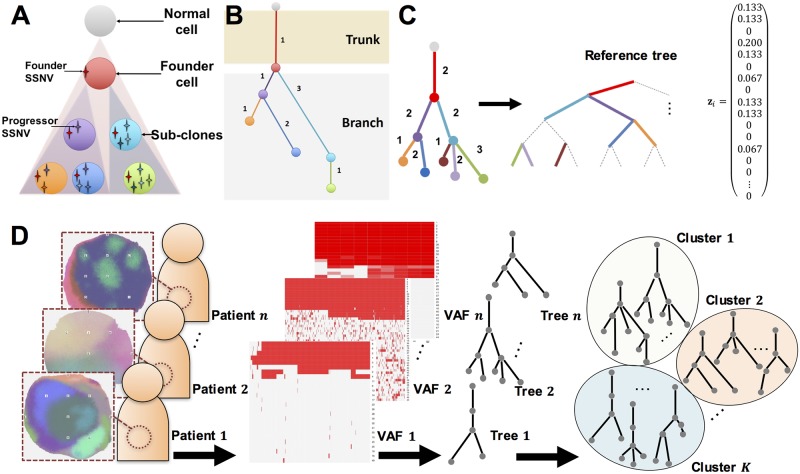
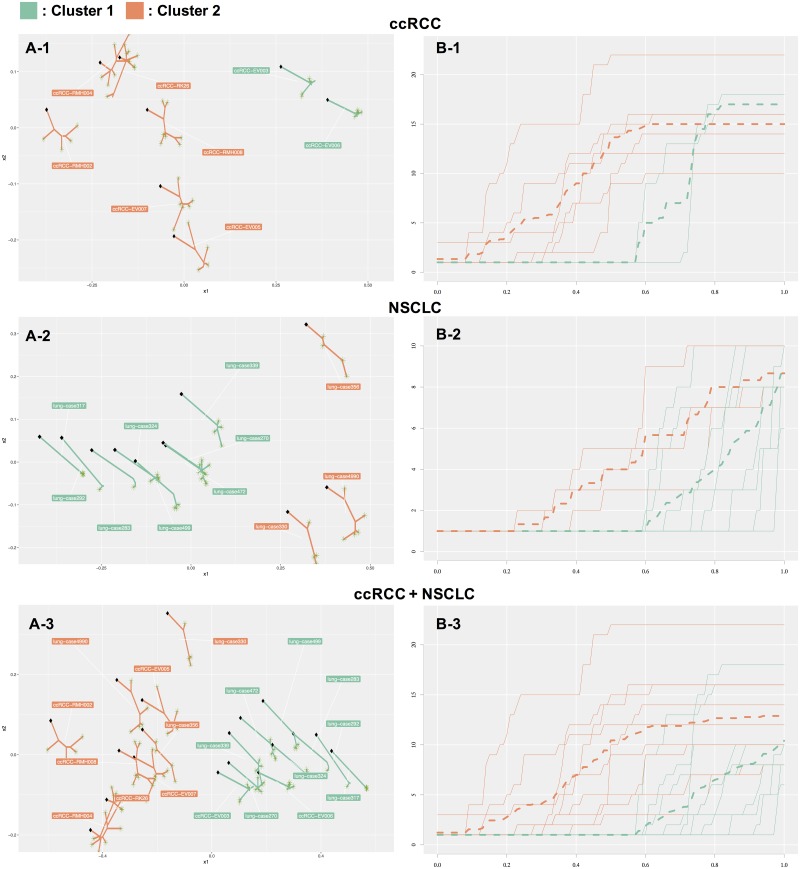
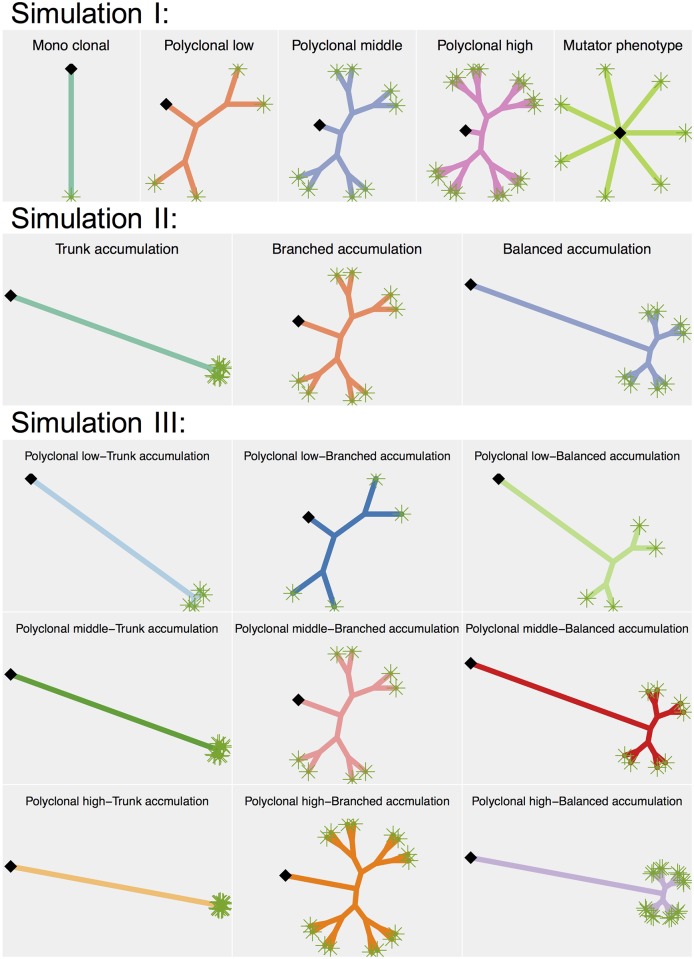
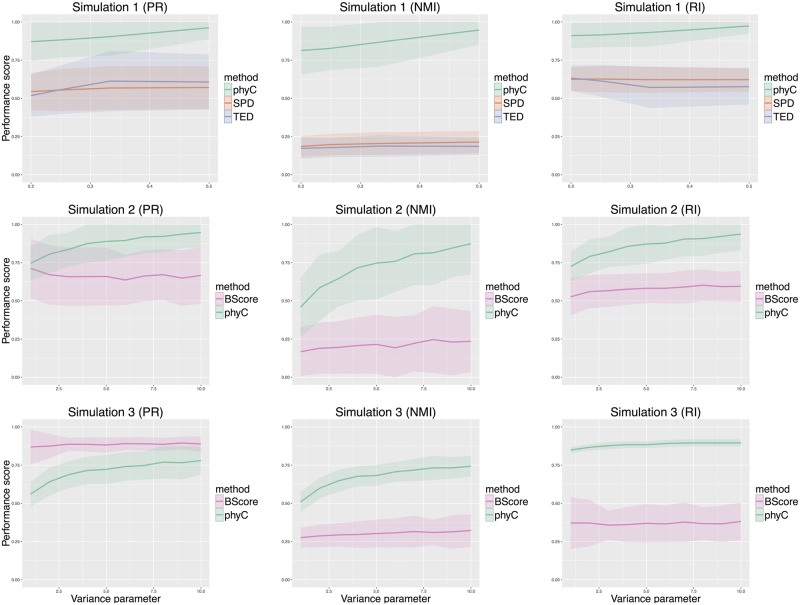
Multi-regional sequencing provides new opportunities to investigate genetic heterogeneity within or between common tumors from an evolutionary perspective. Several state-of-the-art methods have been proposed for reconstructing cancer evolutionary trees based on multi-regional sequencing data to develop models of cancer evolution. However, there have been few studies on comparisons of a set of cancer evolutionary trees. We propose a clustering method (phyC) for cancer evolutionary trees, in which sub-groups of the trees are identified based on topology and edge length attributes. For interpretation, we also propose a method for evaluating the sub-clonal diversity of trees in the clusters, which provides insight into the acceleration of sub-clonal expansion. Simulation showed that the proposed method can detect true clusters with sufficient accuracy. Application of the method to actual multi-regional sequencing data of clear cell renal carcinoma and non-small cell lung cancer allowed for the detection of clusters related to cancer type or phenotype. phyC is implemented with R(≥3.2.2) and is available from https://github.com/ymatts/phyC.
多区域测序为从进化角度研究常见肿瘤内部或之间的遗传异质性提供了新机会。已经提出了几种基于多区域测序数据重建癌症进化树以建立癌症进化模型的先进方法。然而,关于一组癌症进化树比较的研究很少。我们提出了一种用于癌症进化树的聚类方法(phyC),其中基于拓扑结构和边长度属性识别树的子组。为了便于解释,我们还提出了一种评估聚类中树的亚克隆多样性的方法,该方法为亚克隆扩增的加速提供了见解。模拟表明,所提出的方法能够以足够的准确性检测到真实的聚类。将该方法应用于透明细胞肾细胞癌和非小细胞肺癌的实际多区域测序数据,能够检测到与癌症类型或表型相关的聚类。phyC是用R(≥3.2.2)实现的,可从https://github.com/ymatts/phyC获取。