Sundermann Linda K, Wintersinger Jeff, Rätsch Gunnar, Stoye Jens, Morris Quaid
Donnelly Centre for Cellular and Biomolecular Research, University of Toronto, Toronto, Ontario, Canada.
Vector Institute for Artificial Intelligence, Toronto, Ontario, Canada.
PLoS Comput Biol. 2021 Jan 19;17(1):e1008400. doi: 10.1371/journal.pcbi.1008400. eCollection 2021 Jan.
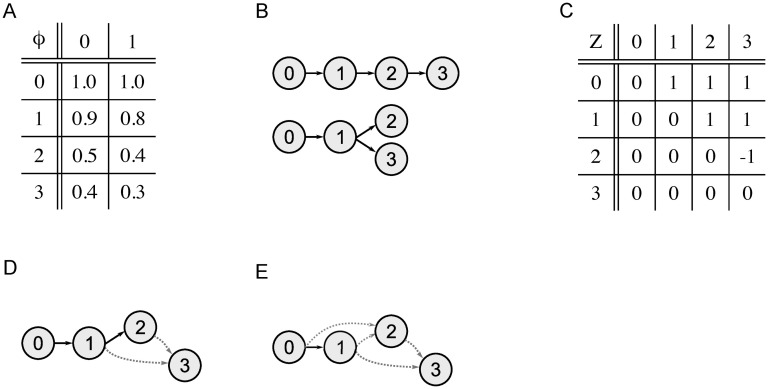
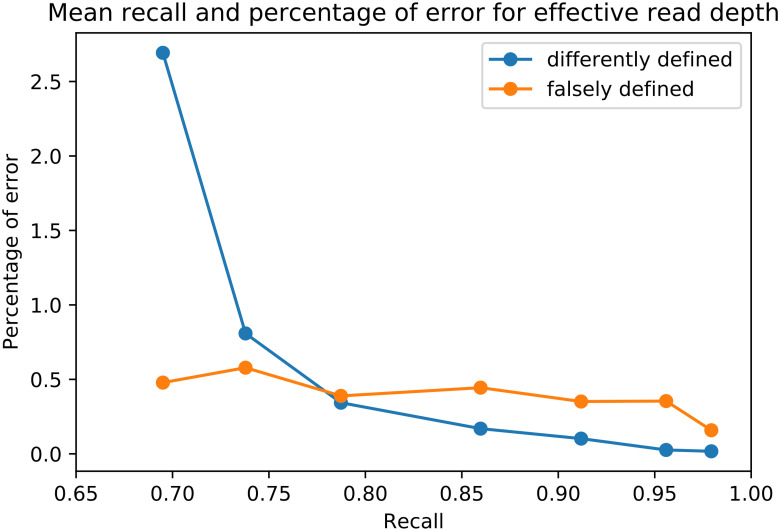
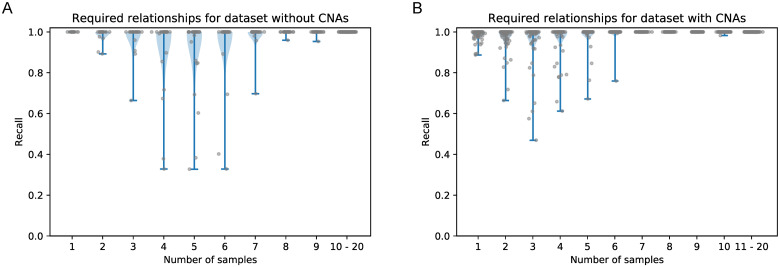
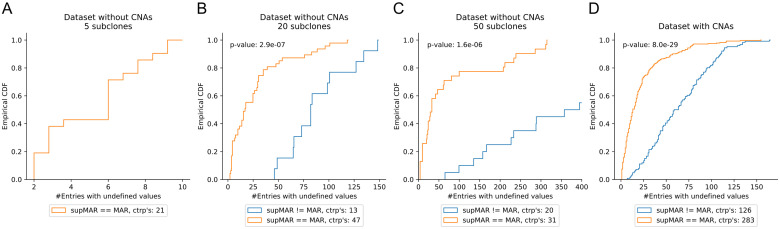
Tumors contain multiple subpopulations of genetically distinct cancer cells. Reconstructing their evolutionary history can improve our understanding of how cancers develop and respond to treatment. Subclonal reconstruction methods cluster mutations into groups that co-occur within the same subpopulations, estimate the frequency of cells belonging to each subpopulation, and infer the ancestral relationships among the subpopulations by constructing a clone tree. However, often multiple clone trees are consistent with the data and current methods do not efficiently capture this uncertainty; nor can these methods scale to clone trees with a large number of subclonal populations. Here, we formalize the notion of a partially-defined clone tree (partial clone tree for short) that defines a subset of the pairwise ancestral relationships in a clone tree, thereby implicitly representing the set of all clone trees that have these defined pairwise relationships. Also, we introduce a special partial clone tree, the Maximally-Constrained Ancestral Reconstruction (MAR), which summarizes all clone trees fitting the input data equally well. Finally, we extend commonly used clone tree validity conditions to apply to partial clone trees and describe SubMARine, a polynomial-time algorithm producing the subMAR, which approximates the MAR and guarantees that its defined relationships are a subset of those present in the MAR. We also extend SubMARine to work with subclonal copy number aberrations and define equivalence constraints for this purpose. Further, we extend SubMARine to permit noise in the estimates of the subclonal frequencies while retaining its validity conditions and guarantees. In contrast to other clone tree reconstruction methods, SubMARine runs in time and space that scale polynomially in the number of subclones. We show through extensive noise-free simulation, a large lung cancer dataset and a prostate cancer dataset that the subMAR equals the MAR in all cases where only a single clone tree exists and that it is a perfect match to the MAR in most of the other cases. Notably, SubMARine runs in less than 70 seconds on a single thread with less than one Gb of memory on all datasets presented in this paper, including ones with 50 nodes in a clone tree. On the real-world data, SubMARine almost perfectly recovers the previously reported trees and identifies minor errors made in the expert-driven reconstructions of those trees. The freely-available open-source code implementing SubMARine can be downloaded at https://github.com/morrislab/submarine.
肿瘤包含多个基因不同的癌细胞亚群。重建它们的进化历史可以增进我们对癌症如何发展以及如何对治疗做出反应的理解。亚克隆重建方法将突变聚类成在同一亚群中共同出现的组,估计属于每个亚群的细胞频率,并通过构建克隆树来推断亚群之间的祖先关系。然而,通常多个克隆树与数据一致,而当前方法无法有效地捕捉这种不确定性;这些方法也无法扩展到具有大量亚克隆群体的克隆树。在这里,我们形式化了部分定义的克隆树(简称为部分克隆树)的概念,它定义了克隆树中成对祖先关系的一个子集,从而隐式地表示具有这些定义的成对关系的所有克隆树的集合。此外,我们引入了一种特殊的部分克隆树,即最大约束祖先重建(MAR),它总结了所有同样适合输入数据的克隆树。最后,我们将常用的克隆树有效性条件扩展到适用于部分克隆树,并描述了SubMARine,这是一种产生subMAR的多项式时间算法,它近似于MAR并保证其定义的关系是MAR中存在的关系的一个子集。我们还扩展了SubMARine以处理亚克隆拷贝数变异,并为此定义了等价约束。此外,我们扩展了SubMARine以允许亚克隆频率估计中的噪声,同时保留其有效性条件和保证。与其他克隆树重建方法相比,SubMARine在时间和空间上的运行与亚克隆数量成多项式比例。我们通过广泛的无噪声模拟、一个大型肺癌数据集和一个前列腺癌数据集表明,在只有单个克隆树存在的所有情况下,subMAR等于MAR,并且在大多数其他情况下它与MAR完美匹配。值得注意的是,在本文呈现的所有数据集上,包括克隆树中有50个节点的数据集,SubMARine在单线程上运行不到70秒,内存使用不到1GB。在实际数据上,SubMARine几乎完美地恢复了先前报告的树,并识别出在专家驱动的这些树的重建中所犯的小错误。实现SubMARine的免费开源代码可在https://github.com/morrislab/submarine下载。