Section of Gastroenterology, Hepatology and Nutrition, Department of Medicine, University of Chicago Medicine, Chicago, IL, USA.
Present address: Boston Children's Hospital, Inflammatory Bowel Disease Center, Boston, MA, USA.
Microbiome. 2017 May 4;5(1):50. doi: 10.1186/s40168-017-0270-x.
Fecal microbiota transplantation (FMT) is an effective treatment for recurrent Clostridium difficile infection and shows promise for treating other medical conditions associated with intestinal dysbioses. However, we lack a sufficient understanding of which microbial populations successfully colonize the recipient gut, and the widely used approaches to study the microbial ecology of FMT experiments fail to provide enough resolution to identify populations that are likely responsible for FMT-derived benefits.
We used shotgun metagenomics together with assembly and binning strategies to reconstruct metagenome-assembled genomes (MAGs) from fecal samples of a single FMT donor. We then used metagenomic mapping to track the occurrence and distribution patterns of donor MAGs in two FMT recipients.
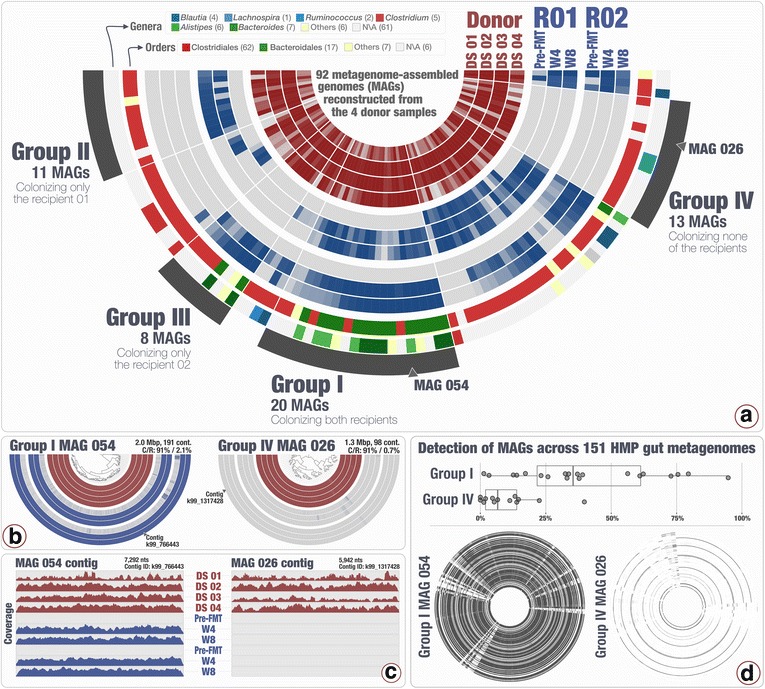
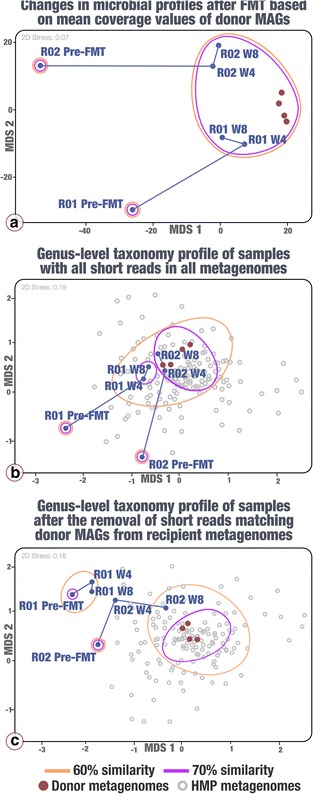
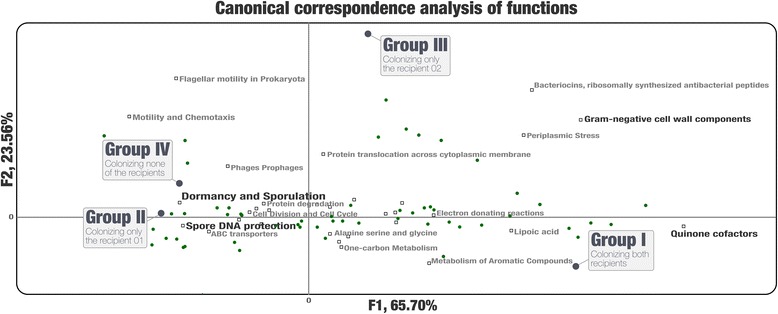
Our analyses revealed that 22% of the 92 highly complete bacterial MAGs that we identified from the donor successfully colonized and remained abundant in two recipients for at least 8 weeks. Most MAGs with a high colonization rate belonged to the order Bacteroidales. The vast majority of those that lacked evidence of colonization belonged to the order Clostridiales, and colonization success was negatively correlated with the number of genes related to sporulation. Our analysis of 151 publicly available gut metagenomes showed that the donor MAGs that colonized both recipients were prevalent, and the ones that colonized neither were rare across the participants of the Human Microbiome Project. Although our dataset showed a link between taxonomy and the colonization ability of a given MAG, we also identified MAGs that belong to the same taxon with different colonization properties, highlighting the importance of an appropriate level of resolution to explore the functional basis of colonization and to identify targets for cultivation, hypothesis generation, and testing in model systems.
The analytical strategy adopted in our study can provide genomic insights into bacterial populations that may be critical to the efficacy of FMT due to their success in gut colonization and metabolic properties, and guide cultivation efforts to investigate mechanistic underpinnings of this procedure beyond associations.
粪便微生物群移植(FMT)是治疗复发性艰难梭菌感染的有效方法,并且在治疗其他与肠道菌群失调相关的医学病症方面显示出前景。然而,我们对哪些微生物群体成功定植于受者肠道缺乏足够的了解,而广泛用于研究 FMT 实验微生物生态学的方法无法提供足够的分辨率来识别可能导致 FMT 获益的群体。
我们使用鸟枪法宏基因组学以及组装和分类策略,从单个 FMT 供体的粪便样本中重建宏基因组组装基因组(MAG)。然后,我们使用宏基因组映射来跟踪供体 MAG 在两名 FMT 受者中的出现和分布模式。
我们的分析表明,我们从供体中鉴定的 92 个高度完整的细菌 MAG 中有 22%成功定植,并在两名受者中至少 8 周保持丰富。具有高定植率的大多数 MAG 属于拟杆菌目。绝大多数缺乏定植证据的 MAG 属于梭菌目,而定植成功率与与孢子形成相关的基因数量呈负相关。我们对 151 个公开可用的肠道宏基因组的分析表明,定植于两个受者的供体 MAG 普遍存在,而在人类微生物组计划的参与者中,既未定植于一个受者也未定植于两个受者的 MAG 则很少见。虽然我们的数据集显示了分类与特定 MAG 定植能力之间的联系,但我们也鉴定了属于同一分类群但具有不同定植特性的 MAG,这突出了适当分辨率的重要性,以探索定植的功能基础,并在模型系统中识别培养、假设生成和测试的目标。
我们研究中采用的分析策略可以为由于其在肠道定植和代谢特性方面的成功而可能对 FMT 疗效至关重要的细菌群体提供基因组见解,并指导培养工作,以调查该程序的机制基础,超越关联进行假设生成和测试。