Bowerman Samuel, Rana Ambar S J B, Rice Amy, Pham Grace H, Strieter Eric R, Wereszczynski Jeff
Department of Physics and Center for the Molecular Study of Condensed Soft Matter, Illinois Institute of Technology , Chicago, Illinois 60616, United States.
Department of Chemistry, University of Massachusetts-Amherst , Amherst, Massachusetts 01003, United States.
J Chem Theory Comput. 2017 Jun 13;13(6):2418-2429. doi: 10.1021/acs.jctc.7b00059. Epub 2017 May 17.
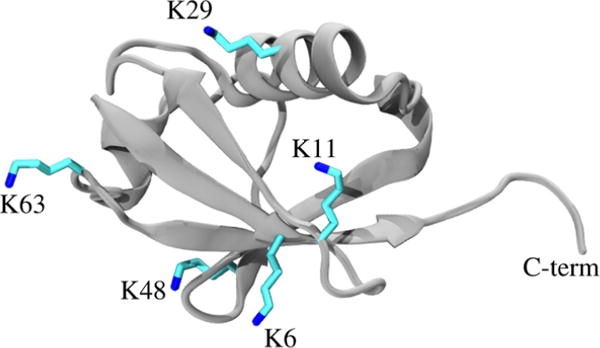
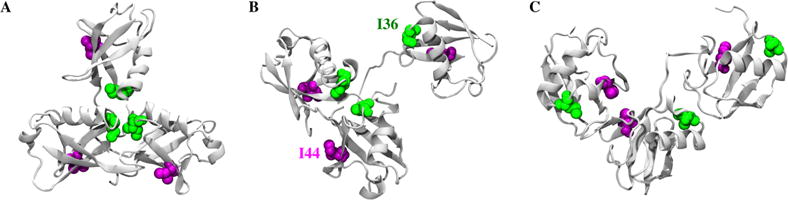
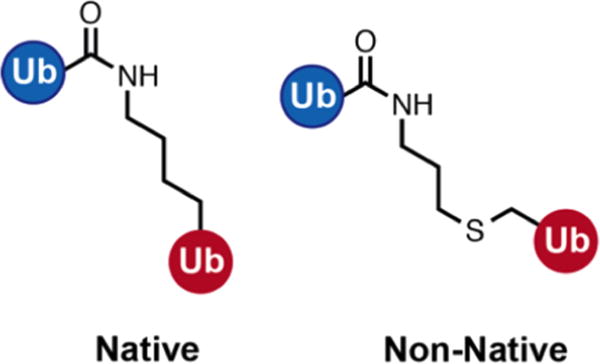
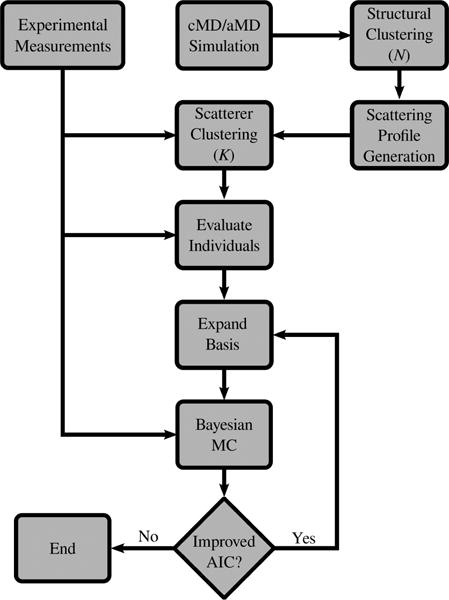
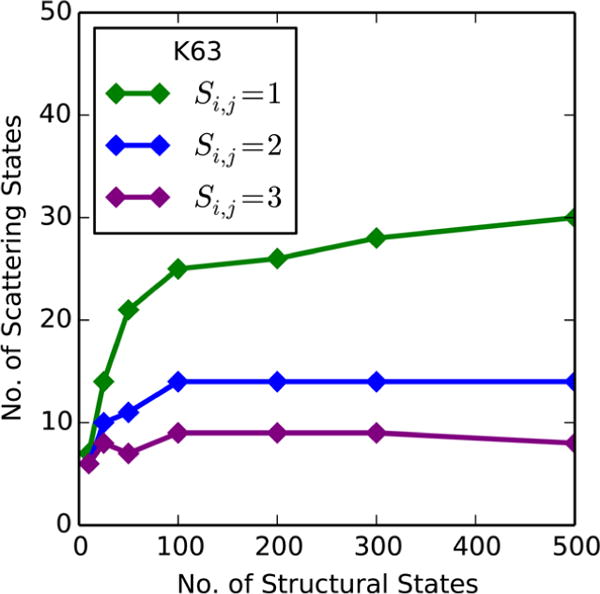
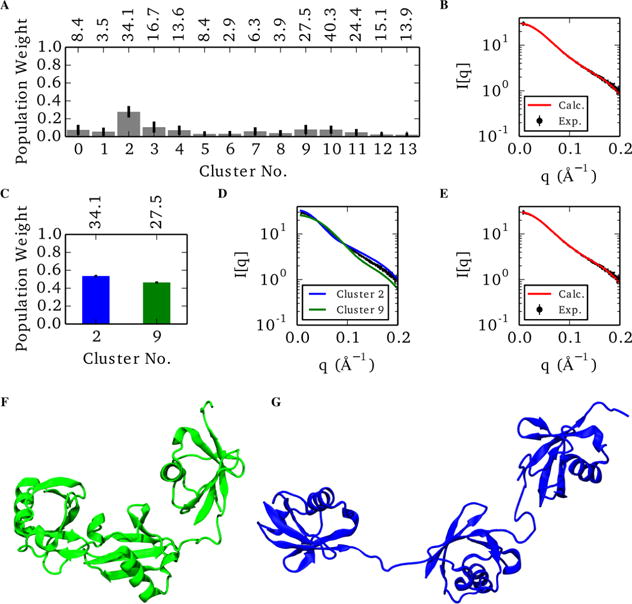
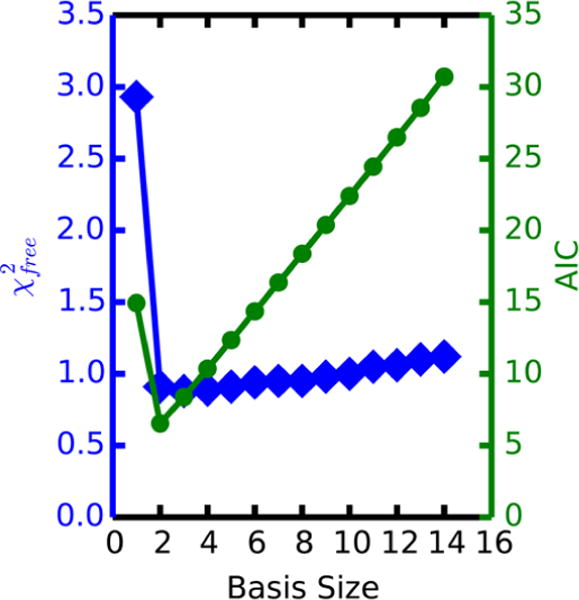
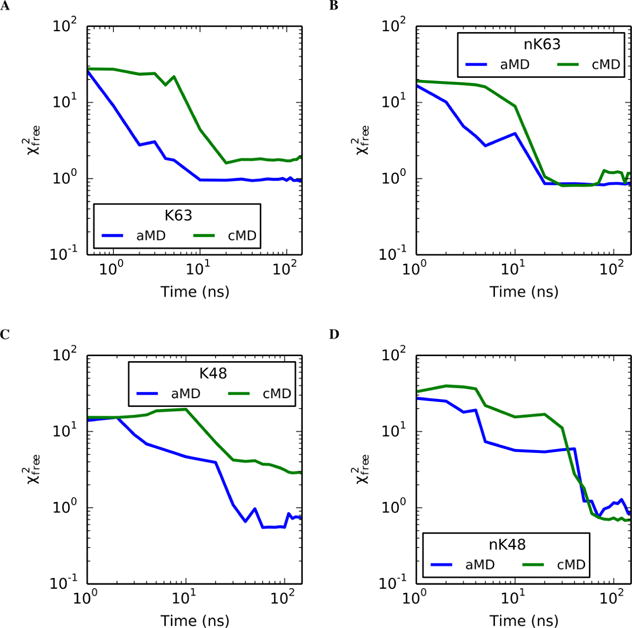
Small-angle X-ray scattering (SAXS) has become an increasingly popular technique for characterizing the solution ensemble of flexible biomolecules. However, data resulting from SAXS is typically low-dimensional and is therefore difficult to interpret without additional structural knowledge. In theory, molecular dynamics (MD) trajectories can provide this information, but conventional simulations rarely sample the complete ensemble. Here, we demonstrate that accelerated MD simulations can be used to produce higher quality models in shorter time scales than standard simulations, and we present an iterative Bayesian Monte Carlo method that is able to identify multistate ensembles without overfitting. This methodology is applied to several ubiquitin trimers to demonstrate the effect of linkage type on the solution states of the signaling protein. We observe that the linkage site directly affects the solution flexibility of the trimer and theorize that this difference in plasticity contributes to their disparate roles in vivo.
小角X射线散射(SAXS)已成为一种越来越流行的用于表征柔性生物分子溶液系综的技术。然而,SAXS产生的数据通常是低维的,因此在没有额外结构知识的情况下很难解释。理论上,分子动力学(MD)轨迹可以提供这些信息,但传统模拟很少能对整个系综进行采样。在这里,我们证明了加速MD模拟可用于在比标准模拟更短的时间尺度上生成更高质量的模型,并且我们提出了一种迭代贝叶斯蒙特卡罗方法,该方法能够识别多状态系综而不会过度拟合。这种方法应用于几个泛素三聚体,以证明连接类型对信号蛋白溶液状态的影响。我们观察到连接位点直接影响三聚体的溶液灵活性,并推测这种可塑性差异有助于它们在体内发挥不同的作用。