Solowska Joanna M, Rao Anand N, Baas Peter W
Department of Neurobiology and Anatomy, Drexel University College of Medicine, Philadelphia, PA 19129.
Department of Neurobiology and Anatomy, Drexel University College of Medicine, Philadelphia, PA 19129
Mol Biol Cell. 2017 Jul 1;28(13):1728-1737. doi: 10.1091/mbc.E17-01-0047. Epub 2017 May 11.
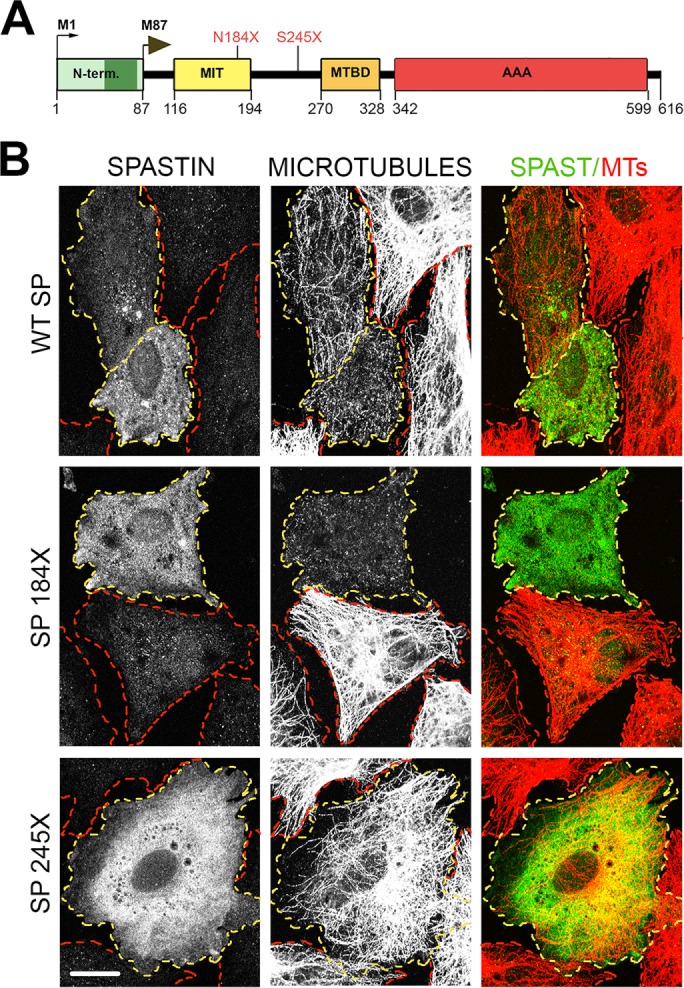
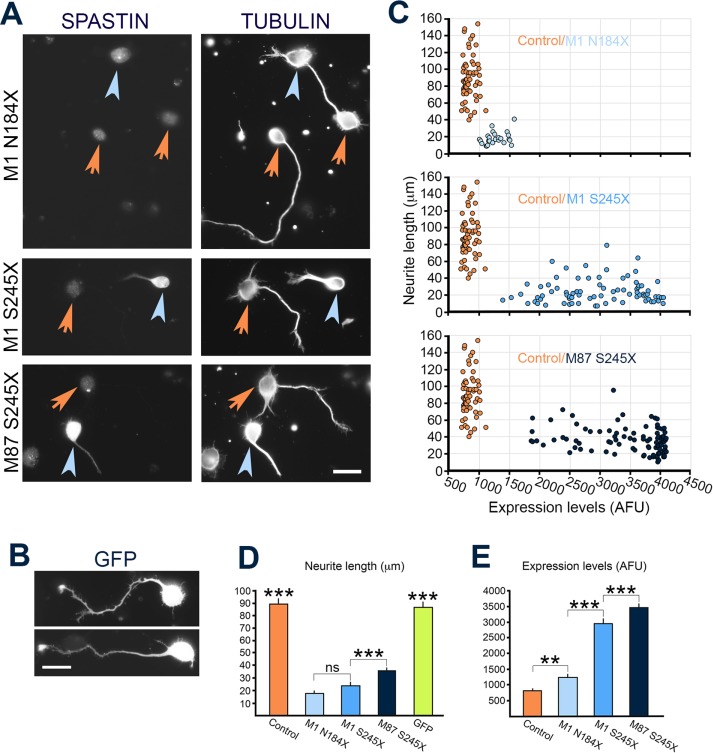
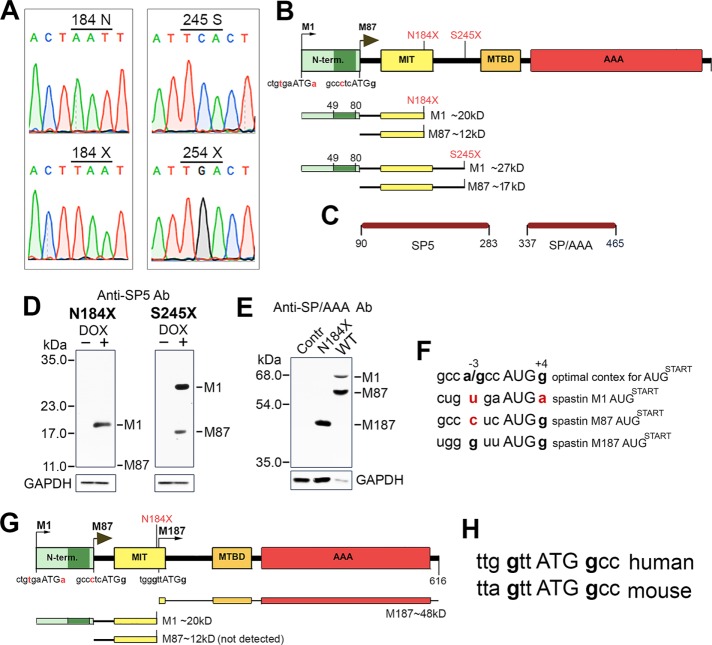
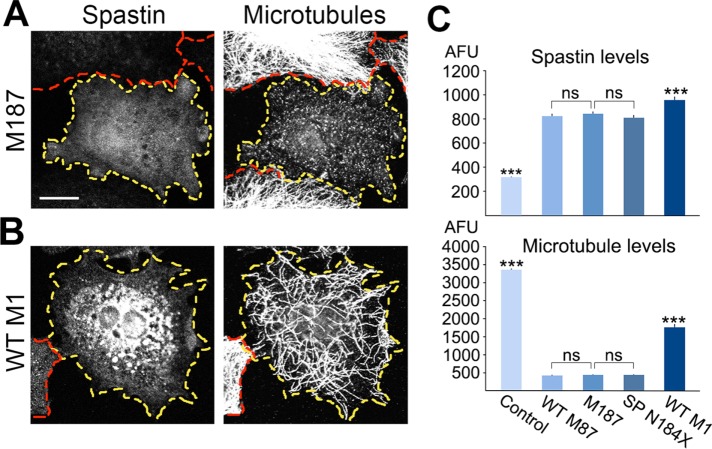
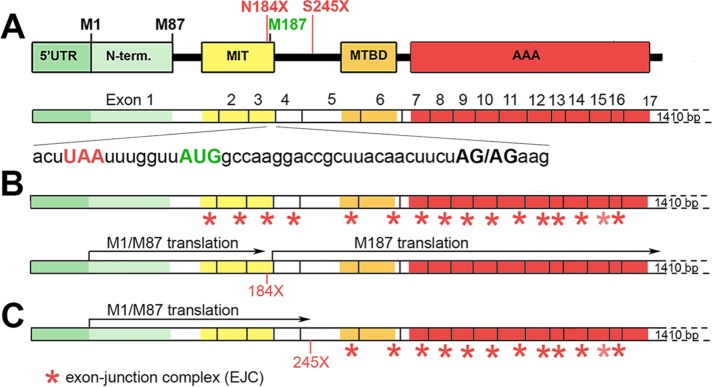
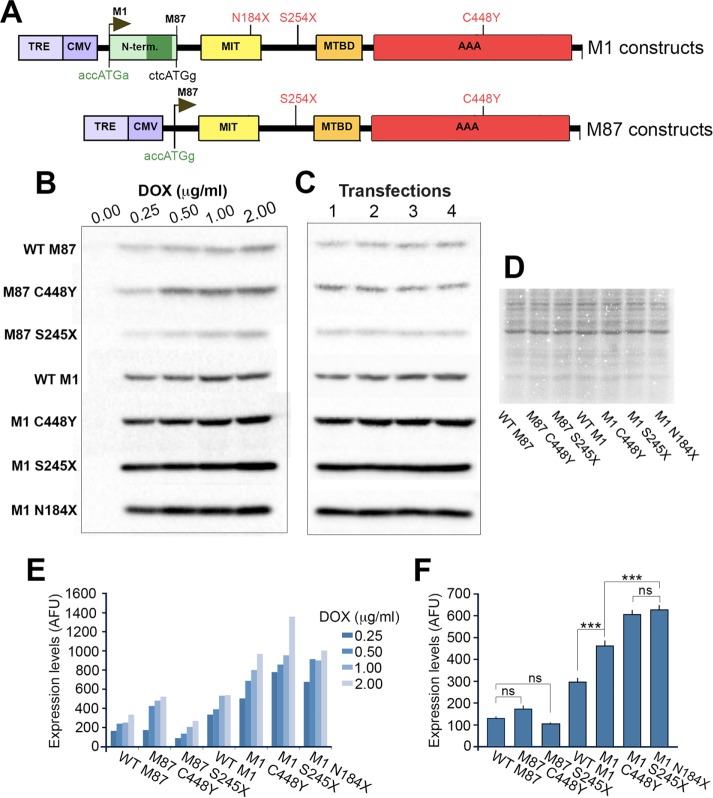
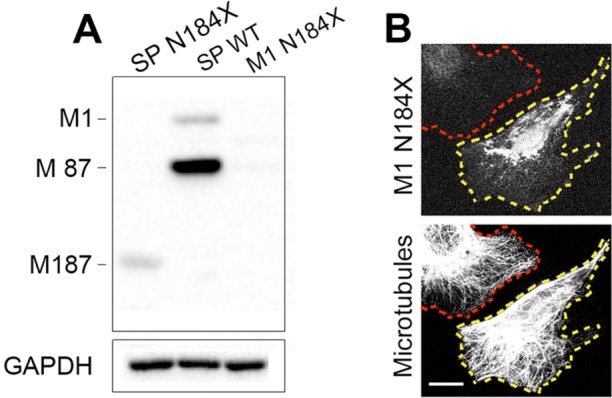
The gene, which produces two isoforms (M1 and M87) of the microtubule-severing protein spastin, is the chief gene mutated in hereditary spastic paraplegia. Haploinsufficiency is a popular explanation for the disease, in part because most of the >200 pathogenic mutations of the gene are truncating and expected to produce only vanishingly small amounts of shortened proteins. Here we studied two such mutations, N184X and S245X, and our results suggest another possibility. We found that the truncated M1 proteins can accumulate to notably higher levels than their truncated M87 or wild-type counterparts. Reminiscent of our earlier studies on a pathogenic mutation that generates full-length M1 and M87 proteins, truncated M1 was notably more detrimental to neurite outgrowth than truncated M87, and this was true for both N184X and S245X. The greater toxicity and tendency to accumulate suggest that, over time, truncated M1 could damage the corticospinal tracts of human patients. Curiously, the N184X mutation triggers the reinitiation of translation at a third start codon in , resulting in synthesis of a novel M187 spastin isoform that is able to sever microtubules. Thus microtubule severing may not be as reduced as previously assumed in the case of that mutation.
该基因可产生微管切断蛋白痉挛素的两种异构体(M1和M87),是遗传性痉挛性截瘫中发生突变的主要基因。单倍剂量不足是对该疾病的一种常见解释,部分原因是该基因的200多种致病性突变大多是截短突变,预计只会产生极少量的缩短蛋白。在这里,我们研究了两个这样的突变,即N184X和S245X,我们的结果提示了另一种可能性。我们发现,截短的M1蛋白比其截短的M87或野生型对应物能积累到显著更高的水平。这让人想起我们早期对一个产生全长M1和M87蛋白的致病性突变的研究,截短的M1对神经突生长的损害明显大于截短的M87,N184X和S245X都是如此。更大的毒性和积累倾向表明,随着时间的推移,截短的M1可能会损害人类患者的皮质脊髓束。奇怪的是,N184X突变会触发在第三个起始密码子处重新开始翻译,从而合成一种能够切断微管的新型M187痉挛素异构体。因此,在该突变的情况下,微管切断可能不像之前假设的那样减少。