Lee Chol Gyu, Iida Toshiya, Inoue Yasuhiro, Muramoto Yasunori, Watanabe Hideki, Nakaho Kazuhiro, Ohkuma Moriya
Japan Collection of Microorganisms, RIKEN BioResource Center.
Central Region Agricultural Research Center, National Agriculture and Food Research Organization.
Microbes Environ. 2017 Jun 24;32(2):118-124. doi: 10.1264/jsme2.ME16136. Epub 2017 May 13.
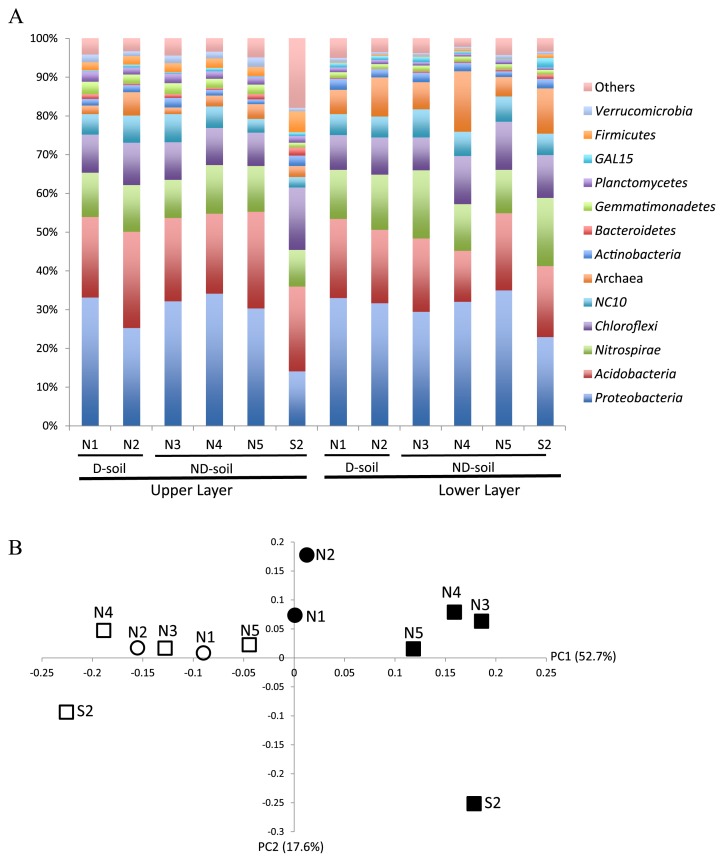
The characterization of microbial communities that promote or suppress soil-borne pathogens is important for controlling plant diseases. We compared prokaryotic communities in soil with or without the signs of tomato bacterial wilt caused by Ralstonia solanacearum. Soil samples were collected from a greenhouse at two different depths because this pathogen is present in deep soil. We used samples from sites in which we detected phcA, a key gene regulating R. solanacearum pathogenicity. The pyrosequencing of prokaryotic 16S rRNA sequences in four soil samples without disease symptoms but with phcA and in two soil samples with disease symptoms indicated that community richness was not significantly different between these two soils; however, microbial diversity in the lower soil layer was higher in soil samples without disease symptoms but with phcA. A difference in prokaryotic community structures between soil samples with and without bacterial wilt was only observed in the upper soil layer despite apparent similarities in the communities at the phylum level. Proteobacteria, Acidobacteria, Chloroflexi, Verrucomicrobia, and several Archaea were more abundant in soil samples without disease symptoms, whereas taxa in another eight phyla were more abundant in soil samples with disease symptoms. Furthermore, some prokaryotic taxa were abundant specifically in the lower layer of soil, regardless of whether disease was present. These prokaryotic taxa may suppress or accelerate the pathogenesis of bacterial wilt and are good targets for future studies on disease control.
鉴定促进或抑制土传病原菌的微生物群落对于控制植物病害至关重要。我们比较了有或没有由青枯雷尔氏菌引起的番茄青枯病症状的土壤中的原核生物群落。由于这种病原菌存在于深层土壤中,所以从温室中两个不同深度采集了土壤样本。我们使用了来自检测到调控青枯雷尔氏菌致病性的关键基因phcA的位点的样本。对四个无病害症状但含有phcA的土壤样本以及两个有病害症状的土壤样本中的原核生物16S rRNA序列进行焦磷酸测序,结果表明这两种土壤之间的群落丰富度没有显著差异;然而,在无病害症状但含有phcA的土壤样本中,下层土壤中的微生物多样性更高。尽管在门水平上群落有明显相似性,但仅在上层土壤中观察到有和没有青枯病的土壤样本之间原核生物群落结构存在差异。变形菌门、酸杆菌门、绿弯菌门、疣微菌门和几个古菌门在无病害症状的土壤样本中更为丰富,而另外八个门中的分类群在有病害症状的土壤样本中更为丰富。此外,一些原核生物分类群无论是否存在病害,都在土壤下层特别丰富。这些原核生物分类群可能抑制或加速青枯病的发病机制,是未来病害控制研究的良好目标。