Vallejos Catalina A, Risso Davide, Scialdone Antonio, Dudoit Sandrine, Marioni John C
MRC Biostatistics Unit, Cambridge Institute of Public Health, Cambridge, UK.
EMBL-European Bioinformatics Institute, Wellcome Genome Campus, Cambridge, UK.
Nat Methods. 2017 Jun;14(6):565-571. doi: 10.1038/nmeth.4292. Epub 2017 May 15.
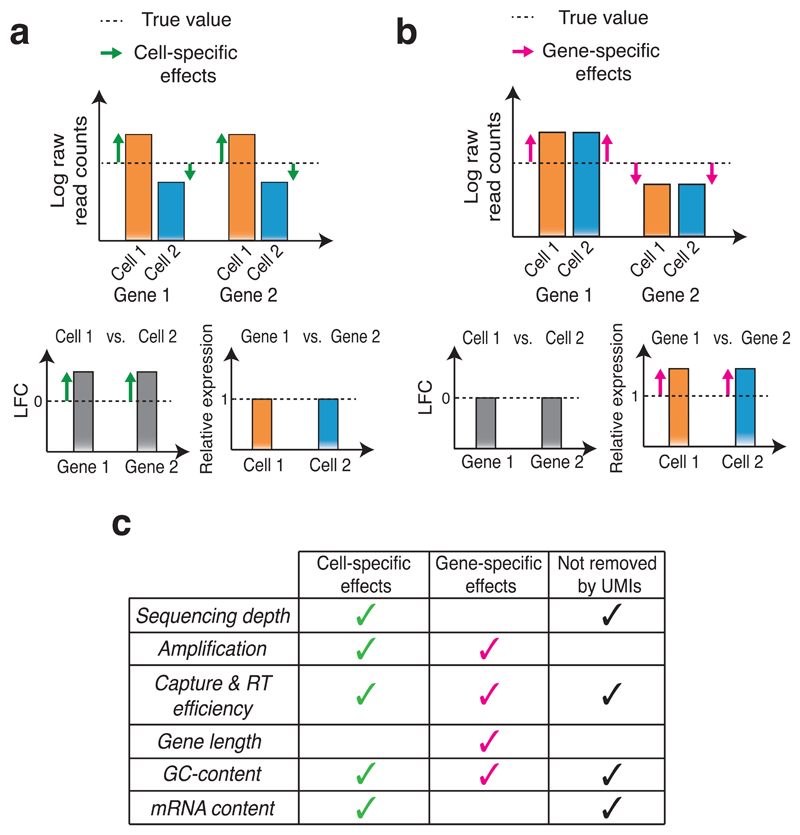
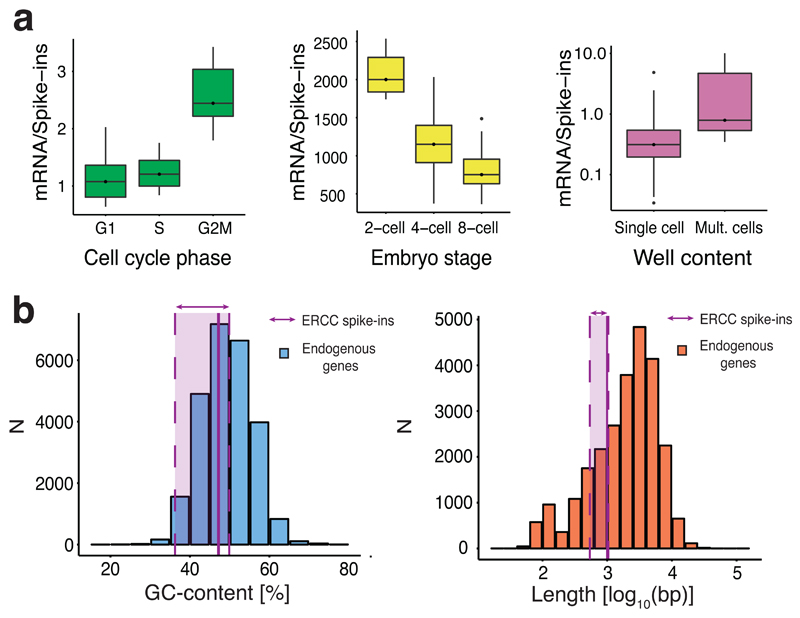
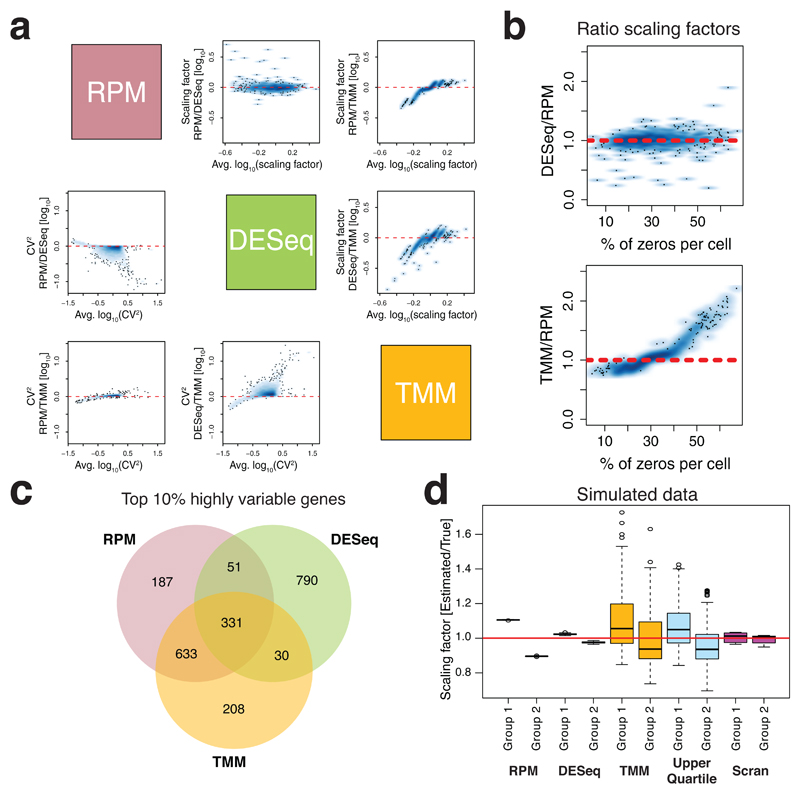
Single-cell transcriptomics is becoming an important component of the molecular biologist's toolkit. A critical step when analyzing data generated using this technology is normalization. However, normalization is typically performed using methods developed for bulk RNA sequencing or even microarray data, and the suitability of these methods for single-cell transcriptomics has not been assessed. We here discuss commonly used normalization approaches and illustrate how these can produce misleading results. Finally, we present alternative approaches and provide recommendations for single-cell RNA sequencing users.
单细胞转录组学正成为分子生物学家工具包的一个重要组成部分。分析使用该技术生成的数据时的一个关键步骤是标准化。然而,标准化通常使用为批量RNA测序甚至微阵列数据开发的方法来进行,并且尚未评估这些方法对单细胞转录组学的适用性。我们在此讨论常用的标准化方法,并说明这些方法如何产生误导性结果。最后,我们提出替代方法,并为单细胞RNA测序用户提供建议。