Lu Songjian, Yang Jiyuan, Yan Lei, Liu Jingjing, Wang Judy Jiaru, Jain Rhea, Yu Jiyang
Department of Computational Biology, St. Jude Children's Research Hospital, Memphis, TN, 38105, USA.
Nat Commun. 2025 Feb 1;16(1):1246. doi: 10.1038/s41467-025-56623-1.
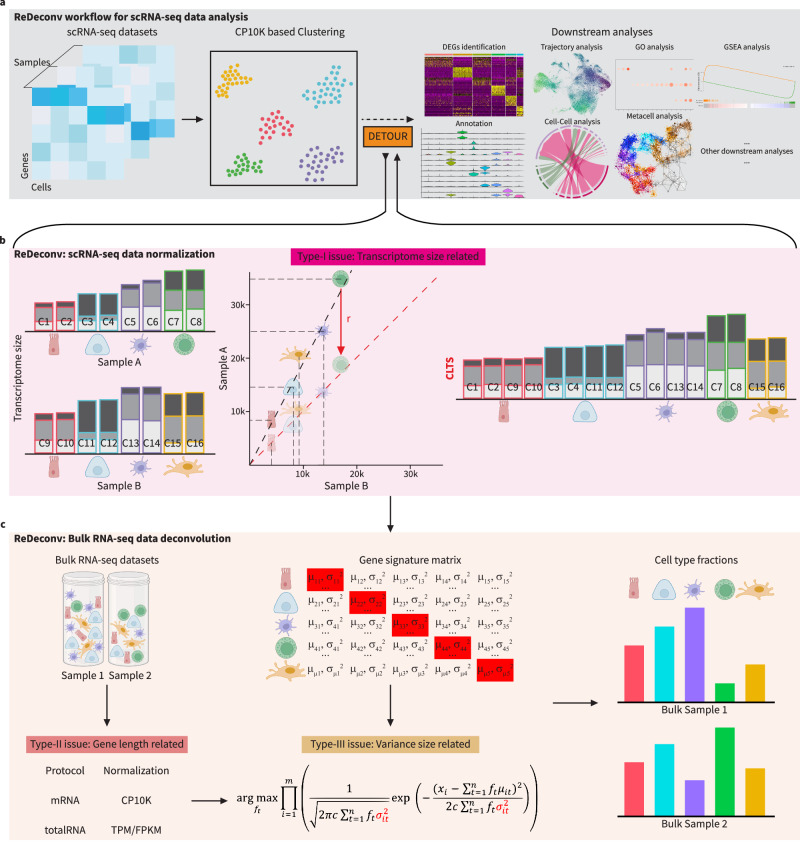
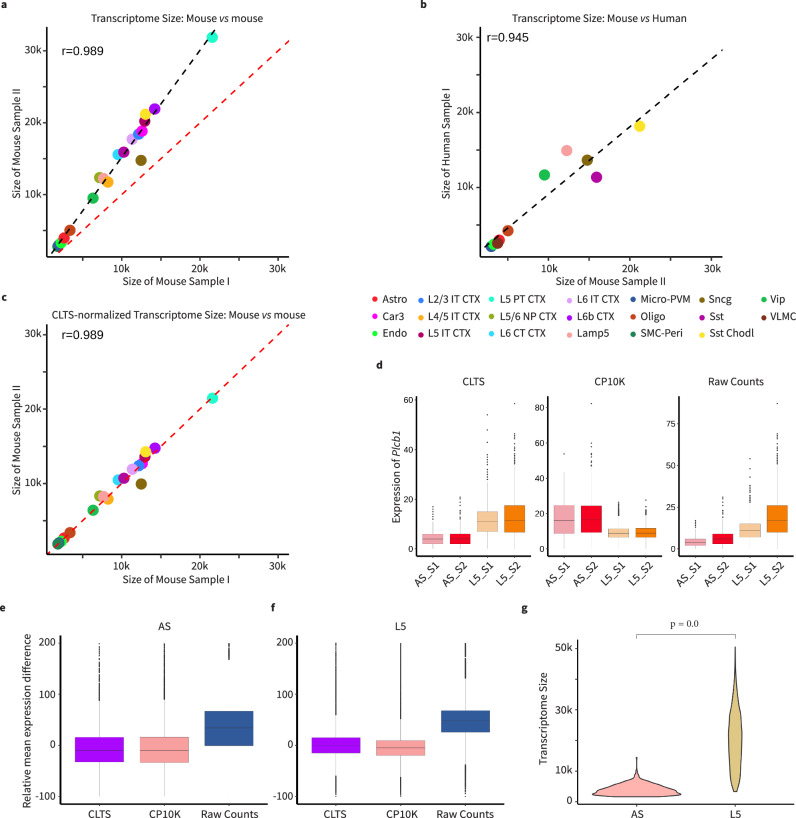
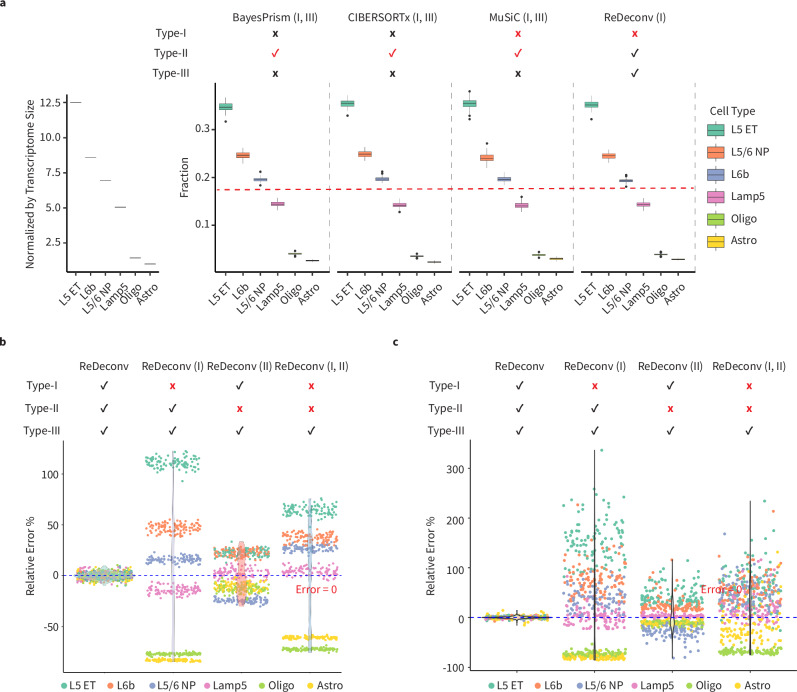
The variation of transcriptome size across cell types significantly impacts single-cell RNA sequencing (scRNA-seq) data normalization and bulk RNA-seq cellular deconvolution, yet this intrinsic feature is often overlooked. Here we introduce ReDeconv, a computational algorithm that incorporates transcriptome size into scRNA-seq normalization and bulk deconvolution. ReDeconv introduces a scRNA-seq normalization approach, Count based on Linearized Transcriptome Size (CLTS), which corrects differential expressed genes typically misidentified by standard count per 10 K normalization, as confirmed by orthogonal validations. By maintaining transcriptome size variation, CLTS-normalized scRNA-seq enhances the accuracy of bulk deconvolution. Additionally, ReDeconv mitigates gene length effects and models expression variances, thereby improving deconvolution outcomes, particularly for rare cell types. Evaluated with both synthetic and real datasets, ReDeconv surpasses existing methods in precision. ReDeconv alters the practice and provides a new standard for scRNA-seq analyses and bulk deconvolution. The software packages and a user-friendly web portal are available.
转录组大小在不同细胞类型间的差异对单细胞RNA测序(scRNA-seq)数据标准化和批量RNA测序细胞反卷积有显著影响,但这一内在特征常被忽视。在此,我们介绍ReDeconv,一种将转录组大小纳入scRNA-seq标准化和批量反卷积的计算算法。ReDeconv引入了一种scRNA-seq标准化方法,即基于线性化转录组大小的计数(CLTS),经正交验证证实,该方法可校正通常在每10K标准化中被误识别的差异表达基因。通过保持转录组大小差异,CLTS标准化的scRNA-seq提高了批量反卷积的准确性。此外,ReDeconv减轻了基因长度效应并对表达方差进行建模,从而改善反卷积结果,特别是对于稀有细胞类型。通过合成数据集和真实数据集评估,ReDeconv在精度上超越了现有方法。ReDeconv改变了实践方式,为scRNA-seq分析和批量反卷积提供了新的标准。软件包和用户友好的门户网站均可获取。