Williams Robert W, Xue Bin, Uversky Vladimir N, Dunker A Keith
Department of Biomedical Informatics; Uniformed Services University; Bethesda, MD USA.
Center for Computational Biology and Bioinformatics; Indiana School of Medicine; Indianapolis, IN USA.
Intrinsically Disord Proteins. 2013 Apr 1;1(1):e25724. doi: 10.4161/idp.25724. eCollection 2013 Jan-Dec.
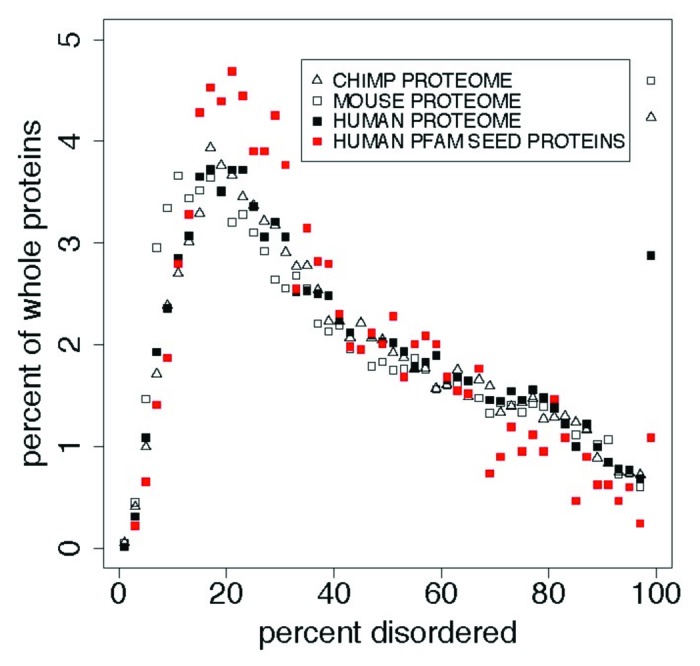
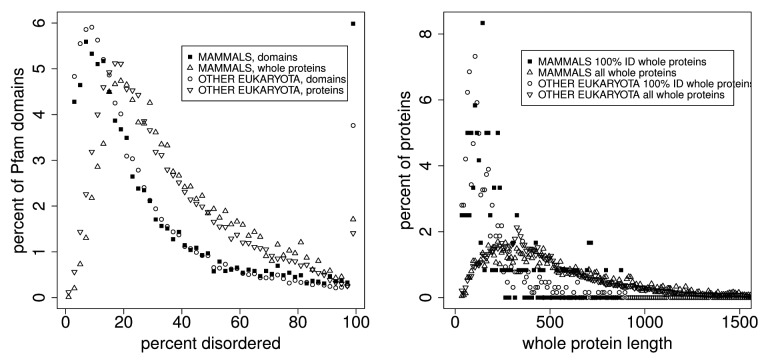
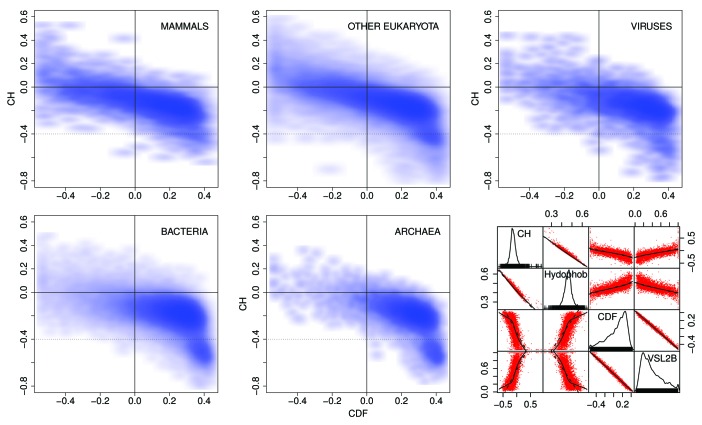
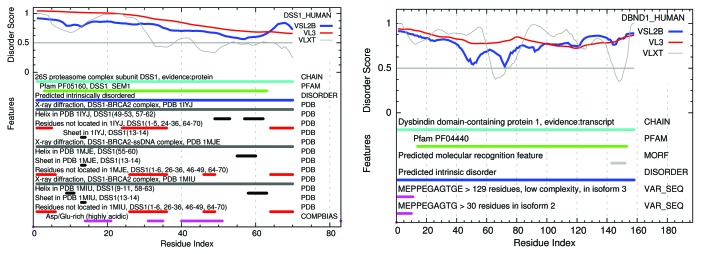
The Pfam database groups regions of proteins by how well hidden Markov models (HMMs) can be trained to recognize similarities among them. Conservation pressure is probably in play here. The Pfam seed training set includes sequence and structure information, being drawn largely from the PDB. A long standing hypothesis among intrinsically disordered protein (IDP) investigators has held that conservation pressures are also at play in the evolution of different kinds of intrinsic disorder, but we find that predicted intrinsic disorder (PID) is not always conserved across Pfam domains. Here we analyze distributions and clusters of PID regions in 193024 members of the version 23.0 Pfam seed database. To include the maximum information available for proteins that remain unfolded in solution, we employ the 10 linearly independent Kidera factors for the amino acids, combined with PONDR predictions of disorder tendency, to transform the sequences of these Pfam members into an 11 column matrix where the number of rows is the length of each Pfam region. Cluster analyses of the set of all regions, including those that are folded, show 6 groupings of domains. Cluster analyses of domains with mean VSL2b scores greater than 0.5 (half predicted disorder or more) show at least 3 separated groups. It is hypothesized that grouping sets into shorter sequences with more uniform length will reveal more information about intrinsic disorder and lead to more finely structured and perhaps more accurate predictions. HMMs could be trained to include this information.
Pfam数据库根据隐马尔可夫模型(HMM)训练识别蛋白质区域间相似性的效果对蛋白质区域进行分组。这里可能存在保守压力。Pfam种子训练集包含序列和结构信息,主要取自蛋白质数据银行(PDB)。在内在无序蛋白质(IDP)研究人员中,长期存在的一种假设认为,保守压力在不同类型内在无序的进化中也起作用,但我们发现预测的内在无序(PID)在Pfam结构域中并非总是保守的。在此,我们分析了Pfam种子数据库23.0版本中193024个成员的PID区域的分布和聚类情况。为了纳入溶液中仍未折叠的蛋白质的最大可用信息,我们使用氨基酸的10个线性独立的基德拉伸因子,结合无序倾向的PONDR预测,将这些Pfam成员的序列转换为一个11列矩阵,其中行数为每个Pfam区域的长度。对所有区域(包括折叠区域)的聚类分析显示有6个结构域分组。对平均VSL2b分数大于0.5(一半或更多为预测无序)的结构域进行聚类分析显示至少有3个分离的组。据推测,将分组集分成长度更均匀的较短序列将揭示更多关于内在无序的信息,并导致更精细的结构以及可能更准确的预测。可以训练HMM来纳入此信息。