Medical School, Division of Basic Sciences, University of Crete, Heraklion 71003, Greece.
Sir William Dunn School of Pathology, University of Oxford, South Parks Road, Oxford OX1 3RE, UK.
Nucleic Acids Res. 2017 Jul 3;45(W1):W300-W306. doi: 10.1093/nar/gkx444.
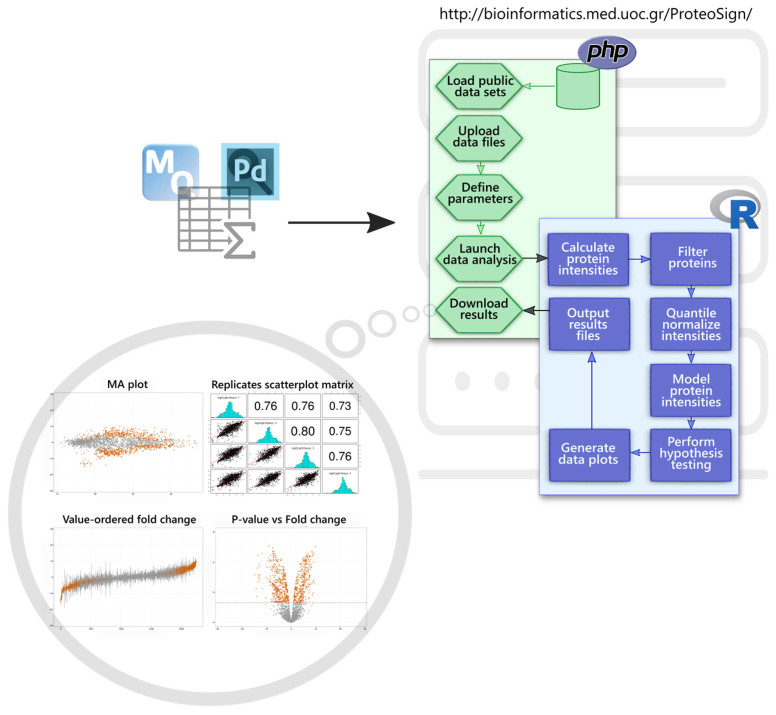
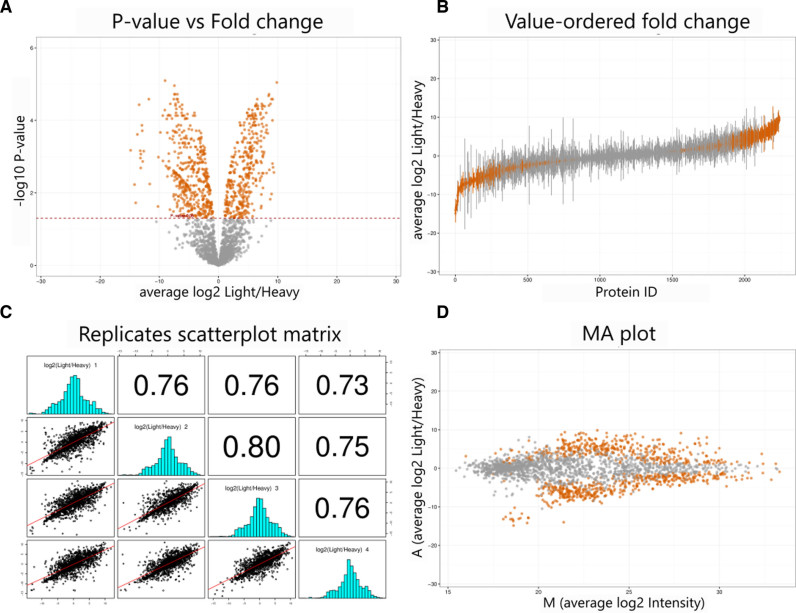
Profiling of proteome dynamics is crucial for understanding cellular behavior in response to intrinsic and extrinsic stimuli and maintenance of homeostasis. Over the last 20 years, mass spectrometry (MS) has emerged as the most powerful tool for large-scale identification and characterization of proteins. Bottom-up proteomics, the most common MS-based proteomics approach, has always been challenging in terms of data management, processing, analysis and visualization, with modern instruments capable of producing several gigabytes of data out of a single experiment. Here, we present ProteoSign, a freely available web application, dedicated in allowing users to perform proteomics differential expression/abundance analysis in a user-friendly and self-explanatory way. Although several non-commercial standalone tools have been developed for post-quantification statistical analysis of proteomics data, most of them are not end-user appealing as they often require very stringent installation of programming environments, third-party software packages and sometimes further scripting or computer programming. To avoid this bottleneck, we have developed a user-friendly software platform accessible via a web interface in order to enable proteomics laboratories and core facilities to statistically analyse quantitative proteomics data sets in a resource-efficient manner. ProteoSign is available at http://bioinformatics.med.uoc.gr/ProteoSign and the source code at https://github.com/yorgodillo/ProteoSign.
蛋白质组动态分析对于了解细胞对内源性和外源性刺激的反应以及维持内稳态至关重要。在过去的 20 年中,质谱(MS)已成为大规模鉴定和描述蛋白质的最强大工具。基于 MS 的蛋白质组学最常见的方法是自上而下的蛋白质组学,在数据管理、处理、分析和可视化方面一直具有挑战性,现代仪器能够从单个实验中产生数千兆字节的数据。在这里,我们介绍了 ProteoSign,这是一个免费的网络应用程序,专门用于允许用户以用户友好和自解释的方式执行蛋白质组学差异表达/丰度分析。尽管已经开发了几个用于蛋白质组学数据定量后统计分析的非商业独立工具,但由于它们通常需要非常严格的编程环境、第三方软件包的安装,有时还需要进一步的脚本编写或计算机编程,因此大多数工具都不受终端用户欢迎。为了避免这种瓶颈,我们开发了一个用户友好的软件平台,可以通过网络界面访问,以便使蛋白质组学实验室和核心设施能够以资源高效的方式对定量蛋白质组学数据集进行统计分析。ProteoSign 可在 http://bioinformatics.med.uoc.gr/ProteoSign 上获得,源代码可在 https://github.com/yorgodillo/ProteoSign 上获得。